
記住我

The results and discussion of this review are organised into sections that detail the use of various radionuclides, as well as summarising findings from in vitro, in vivo and clinical studies relevant to TAT in GB.
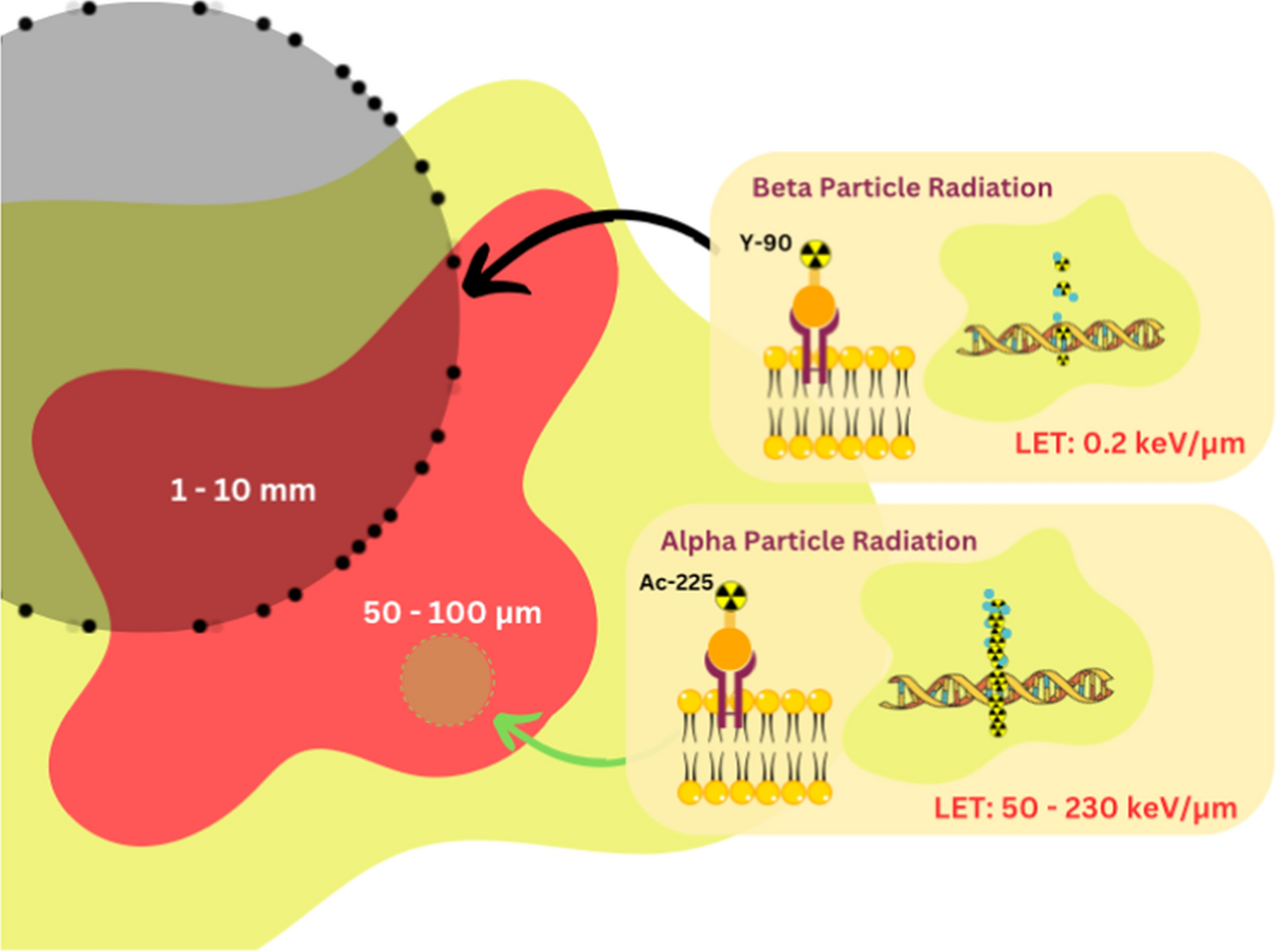
3.1 RadionuclidesA wide array of isotopes has been explored for TAT in the context of GB. Among these isotopes, Astatine-211 (At-211) has been used in 14 pre-clinical studies, Bismuth-213 (Bi-213) in five clinical trials, and Actinium-225 (Ac-225) in six pre-clinical studies and one phase I clinical trial. Each isotope exhibits a unique set of characteristics. Ac-225, characterised by its 100% α-emission, a range of 0.04–0.10 mm, and a half-life (T1/2) of 238.10 h (~ 10 days), is shown to offer several advantages [20,21,22,23,24,25,26,27]. Notably, it exhibits compatibility with DOTA-complexation, rendering it a versatile choice for a variety of compounds [26]. Furthermore, its relatively extended half-life (T1/2) allows for enhanced transport and more efficient distribution before radioactive decay, particularly beneficial when treating larger tumour volumes, as indicated by Cordier et al. [28]. However, the longer T1/2 of Ac-225 results in the generation of multiple alpha particles due to its rapid decay chain (as depicted in Fig. 3). Moreover, recoiled daughters might impact its stability. Studies involving Ac-225 can be found in the references [20,21,22,23,24,25,26].
Fig. 3
Ac-225 decay chain; photons with a branching ratio > 3% relative to 225Ac decay are shown
Bi-213, which boasts a 2.2% α-emission and 97.8% β-emission, possesses a range of 0.05–0.10 mm and a T1/2 of 0.77 h [27,28,29,30,31,32,33]. Like Ac-225, it can be effectively complexed with DOTA, offering a straightforward and universal solution [26, 30,31,32]. However, its short T1/2 and gamma-energy combination make it less efficient in terms of tumour distribution before radioactive decay [28]. The primary drawback of Bi-213 is its brief T1/2, which affects its residence time within critical GB cells. In these cases, the ratio between cell membrane coverage (receptor affinity) and time plays a pivotal role. Studies involving Bi-213 are listed in the references [28,29,30,31,32,33].
Lastly, At-211, featuring 42.0% α-emission and 58.0% electron capture, possesses a range of 0.05 mm and a T1/2 of 7.20 h [27, 34,35,36,37,38,39,40,41,42,43,44,45,46,47]. Its longer T1/2 gives it the potential to have a more advantageous biodistribution. However, At-211 is limited to applications involving mAb and smaller fragments, which can often exhibit low biological and chemical stability. Relevant studies pertaining to 211At are listed in the references [34,35,36,37,38,39,40,41,42,43,44,45,46,47].
3.2 Pre-Clinical StudiesA total of 26 pre-clinical papers were identified that investigated various aspects of TAT for GB. There were 12 in vitro studies, four in vivo studies, nine combined in vitro/in vivo studies, and one modelling study. A summary of pre-clinical studies is presented in Tables 1 and 2.
Table 1 Summary of In Vitro Studies Findings.Table 2 Summary of in vivo study findings3.2.1 In Vitro StudiesIn vitro studies primarily focused on several key aspects, including the binding affinity of different targeting agents, clonogenic survival/cell viability, and the potential for cell cycle arrest.
The binding affinity and binding rate of labelled alpha emitters were consistently high compared to non-targeted and unlabelled emitters. Notably, studies conducted by Zalutsky et al. [39, 41] and Larsen et al. [36] demonstrated that At-211 conjugated to mAbs 81C6/Mel-14 and 2'-deoxyuridine (AUdR) exhibited high binding specificity to targeted cells (ranging from 60.0% to 84.2%) and low binding to non-targeted cells (4.0–5.6%). Moreover, Ma et al. [42] and Liu et al. [43] found that the binding rates to targeted cells were 24.9% and 32.0%, respectively, while the binding rate to non-targeted cells was less than 7%. These studies explored At-221 conjugated to either a heterodimeric peptide (targeting vascular endothelial growth factor receptor (VEGFR) and integrins) or a fibroblast activation protein inhibitor (FAPI).
The therapeutic effectiveness of TAT was assessed in various ways in the literature, including cell viability, relative biological effectiveness (RBE), survival fraction of GB cells at specific absorbed dose rates, and activity concentration resulting in a specific survival rate. Larsen et al. [35, 36] and Rosenkranz et al. [37] investigated the activity concentrations resulting in a 37% survival rate (A37) of GB cells using At-211 conjugated to mAbs 81C6/Mel-14, AUdR, and engineered modular recombinant transporters. The A37 values for conjugated At-211 were significantly lower (4.4–56.6 kBq/ml) compared to free At-211 (32.6–132 kBq/ml). These findings underscore the heightened effectiveness and precision of At-211 conjugated to a targeting agent in killing clonogenic GB cells.
Majkowska-Pilip et al. [25], Ma et al. [42] and Liu et al. [43] reported on cell viability; a dose-dependent decrease in all studies was observed. Ac-225 conjugated to substance-P reduced cell viability to 50% at 50 kBq/ml; At-211 conjugated to FAPI and control reduced viability to 42.1% and 56.5% at 92 kBq/ml, respectively; and At-211 conjugated to heterodimeric peptide and control reduced viability to 47.5% and 62.0% at 75 kBq/ml, respectively. Furthermore, Majkowska-Pilip et al. [25], Ma et al. [42] and Liu et [43] demonstrated significant cell cycle arrest in the G2/M phase in treated cell lines compared to control, with treated cells in G2/M phase ranging from 62.1% to 80% and control cells at 11.9% to 36.6% (cells are more radiosensitive in these phases).
The therapeutic effectiveness of TAT compared to other treatment modalities was explored in several studies. Carlin et al. [38, 40] and Zalutsky et al. [39] highlighted that the 2 Gy survival fraction (SF2) for At-211 was significantly lower than that of Iodine-131 (I-131) and EBRT, meaning that At-211 is much more effective in killing cancer cells when exposed to a 2 Gy dose compared to I-131 and EBRT (i.e., more cytotoxic at this radiation dose). Additionally, Barazznol et al. [48] reported that the RBE of alpha particles compared to x-rays and protons was higher, with RBE10 (calculated at 10% survival) and RBE3Gy (survival level after 3 Gy) being 1.17 and 1.35 for protons and 1.84 and 3.79 for alpha particles, respectively. The RBE is a measure of how effective a particular type of radiation is at causing biological damage, relative to x-rays, although both alpha and protons have a higher RBE than X-rays, alpha particles had the highest RBE, indicating that they are more effective in causing biological damage.
Overall, the pre-clinical in vitro studies on TAT have shown its potential as an effective therapeutic approach for GB treatment. The high binding affinity to targeted cells, dose-dependent reduction in cell viability, and cell cycle arrest and enhanced cytotoxicity compared to other treatments collectively highlight the promising aspects of TAT.
Building on the existing in vitro studies that detail the binding affinity of TAT, cell viability, cell cycle arrest, and other cellular responses in GB treatment, further research is suggested in the following areas to address existing knowledge gaps. This recommendation aligns with the International Atomic Energy Agency (IAEA) [49] guidance for pre-clinical studies with radiopharmaceuticals. Below is an outline of studies that are covered by the included literature and those requiring further investigation:
IAEA Recommended Studies—Covered in Included Literature:
i.Binding affinity and specificity: Several studies have examined the binding affinity and specificity of radiopharmaceuticals like At-211 conjugated with monoclonal antibodies and peptides. For example, Zalutskys and Larsen's [35, 39] studies show significant differences in binding to target versus non-target cells, which aligns with the IAEA's emphasis on understanding binding characteristics.
ii.Cell viability and clonogenic survival: Studies (Table 1) have provided detailed clonogenic survival curves, showing how different doses affect survival rates of GB cells, addressing the IAEA's recommendation for cell viability studies.
iii.Cellular response to radiation: The cellular response to radiation was also explored, including dose-dependent effects and RBE, contributing to understanding the efficacy of different types of radiation as recommended by IAEA.
iv.Therapeutic efficacy: Carlin's [38, 40] studies demonstrated higher cytotoxicity and absorbed dose effectiveness of At-211 compared to I-131 and external beam radiation, providing crucial data on the therapeutic potential of radiopharmaceuticals.
IAEA Recommended Studies—Require Further Investigation:
i.Internalisation and intracellular/subcellular distribution: Despite detailed binding and viability studies, the internalisation dynamics and subcellular localisation of tracers post-binding are not clearly addressed in the summarised studies. These are crucial for understanding how tracers behave inside cells after binding to their targets.
ii.Blocking studies: Although high binding specificity is demonstrated, there is no clear mention of blocking studies to assess the saturability and specificity of binding beyond competitive interactions. Such studies would help confirm the selectivity and potential off-target effects.
iii.Efflux pump assays and blood-brain barrier permeability: There is a lack of explicit studies on how these radiopharmaceuticals interact with the blood-brain barrier or efflux pumps, which is critical for CNS-targeted therapies like those for GB.
iv.Metabolite analysis: It is important to understand the metabolic pathways and by-products after tracer uptake, which has not been detailed in the summary. This could affect both efficacy and toxicity profiles of the radiopharmaceuticals.
v.Functional/efficacy assays beyond viability: Additional functional assays to evaluate the biochemical pathways affected post-binding could provide deeper insights into the mechanistic effects of TAT agents on GB cells.
By addressing these gaps, further research can enhance the understanding of the comprehensive therapeutic profile of TAT, particularly focusing on areas like tracer internalisation and metabolic processing, which are pivotal for ensuring the safety and effectiveness of treatment strategies for GB.
3.2.2 In Vivo StudiesIn vivo studies primarily focused on three aspects: biodistribution of targeted agents, median survival in treated animals versus control groups, and tumour growth/size reduction. Figure 4 provides a summary of the biodistribution of various targeting agents. It reveals that intravenous delivery of targeted agents resulted in tumour uptake ranging from 0.13% to 20.0% of the total injected dose per gram (%ID/g), while intratumoral delivery demonstrated a significantly higher tumour accumulation of 132.5%ID/g [20, 21, 39, 41,42,43]. Furthermore, intravenous injection groups displayed higher tumour-to-organ ratios, with the most significant uptake observed in the liver, spleen and kidneys (Fig. 4B) [20, 41,42,43]. Ma et al. [42] reported more favourable results concerning the tumour-to-organ ratio with intratumoral injection, including tumour-to-liver, tumour-to-kidney, and tumour-to-blood ratios of 11.13, 6.39, and 14.17, respectively, post-injection.
Fig. 4
Biodistribution of the targeting agent. (A) The tumour accumulation (red arrow indicates the intratumoral injection result). (B) The intravenous injection distribution in tumour and healthy organs
The studies revealed promising results in terms of the median overall survival (OS) of mice treated with TAT. Treated groups demonstrated OS ranging from 14.6 to 41 days, while untreated/control groups ranged from 9 to 23 days [20, 21, 24, 42,43,44]. TAT also exhibited a dose-dependent tumour suppression effect, with doses between 180 and 740 kBq resulting in reduced tumour volume ranging from 48% to 77% [12, 37, 42,
留言 (0)