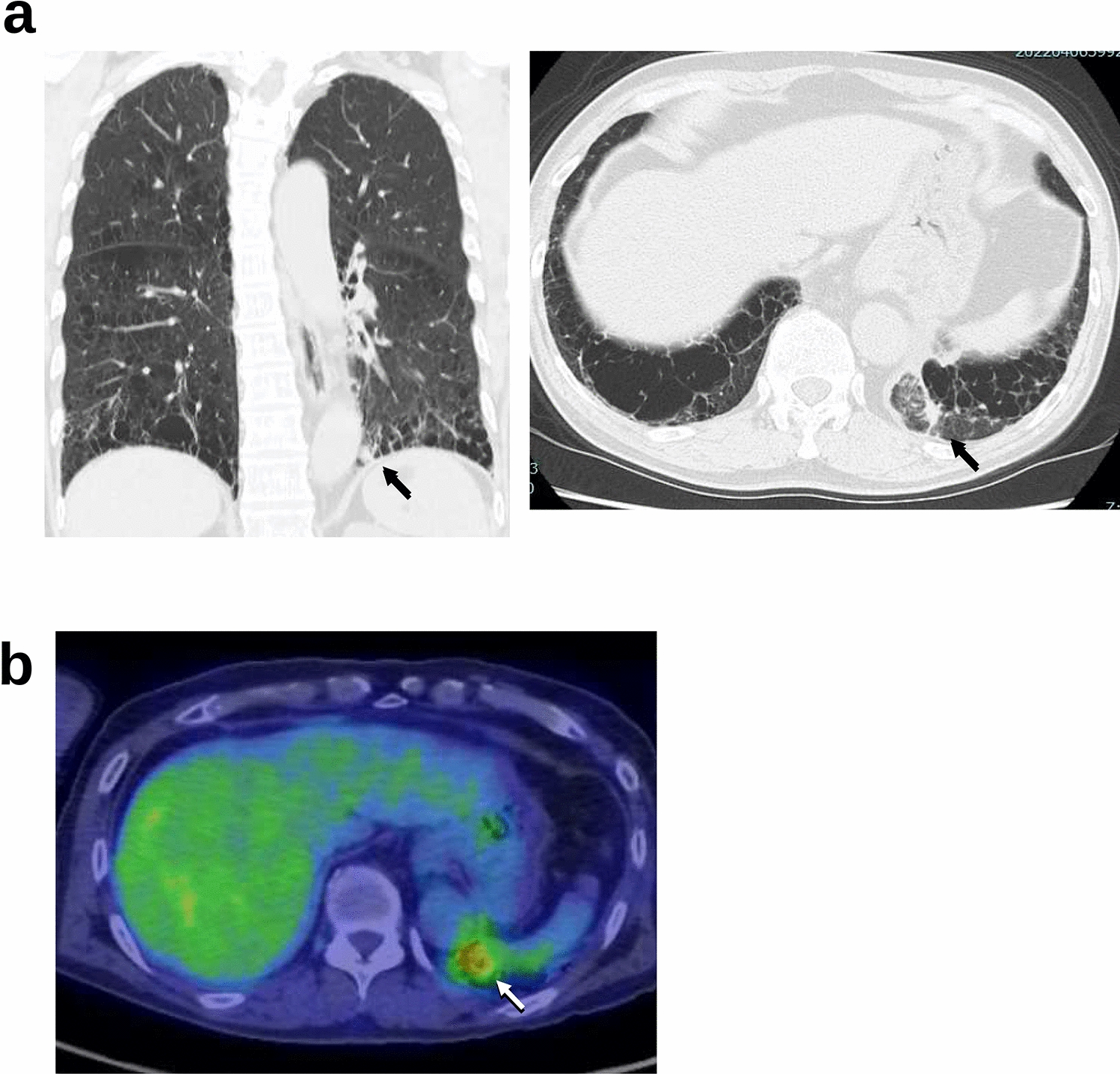
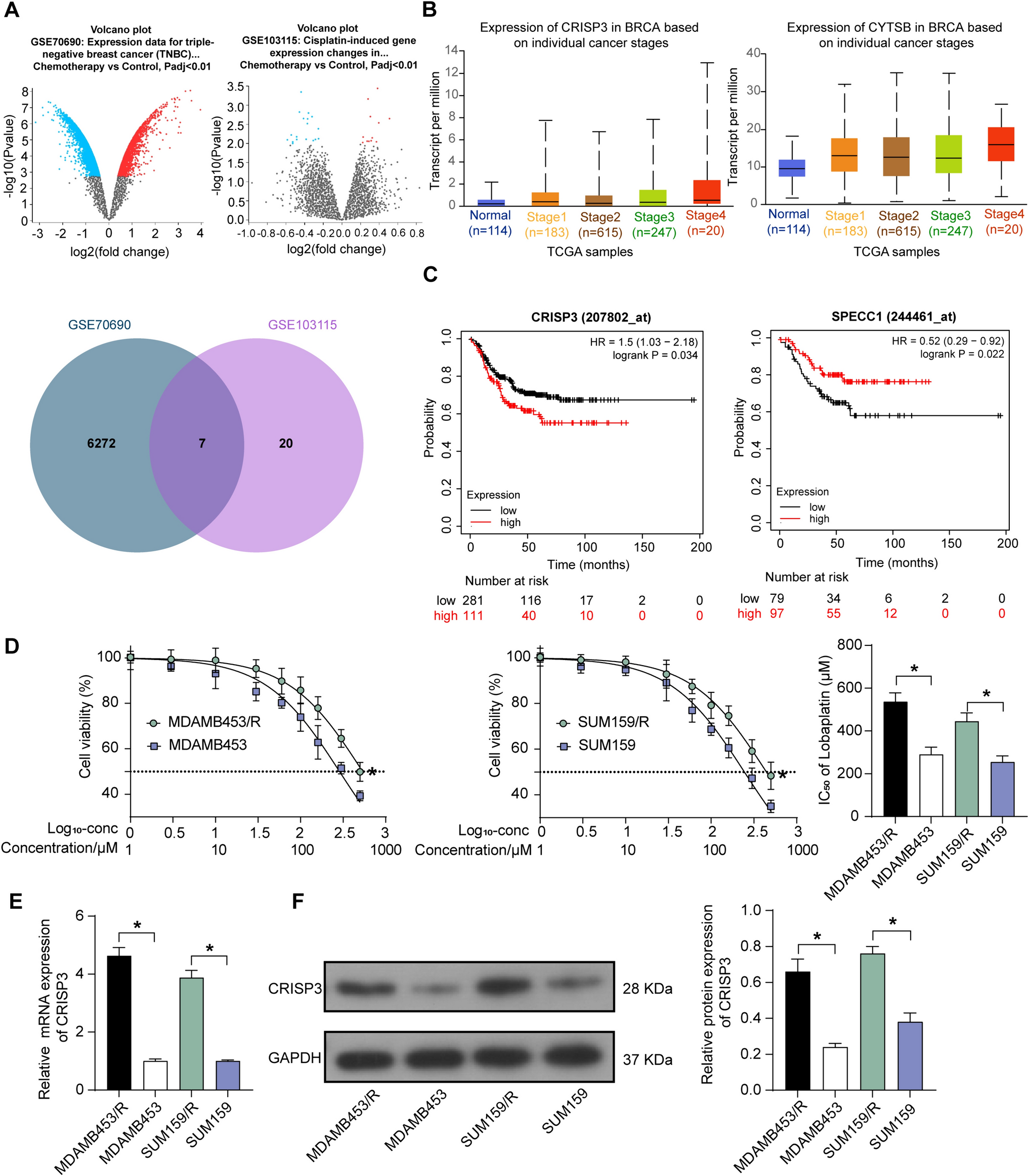
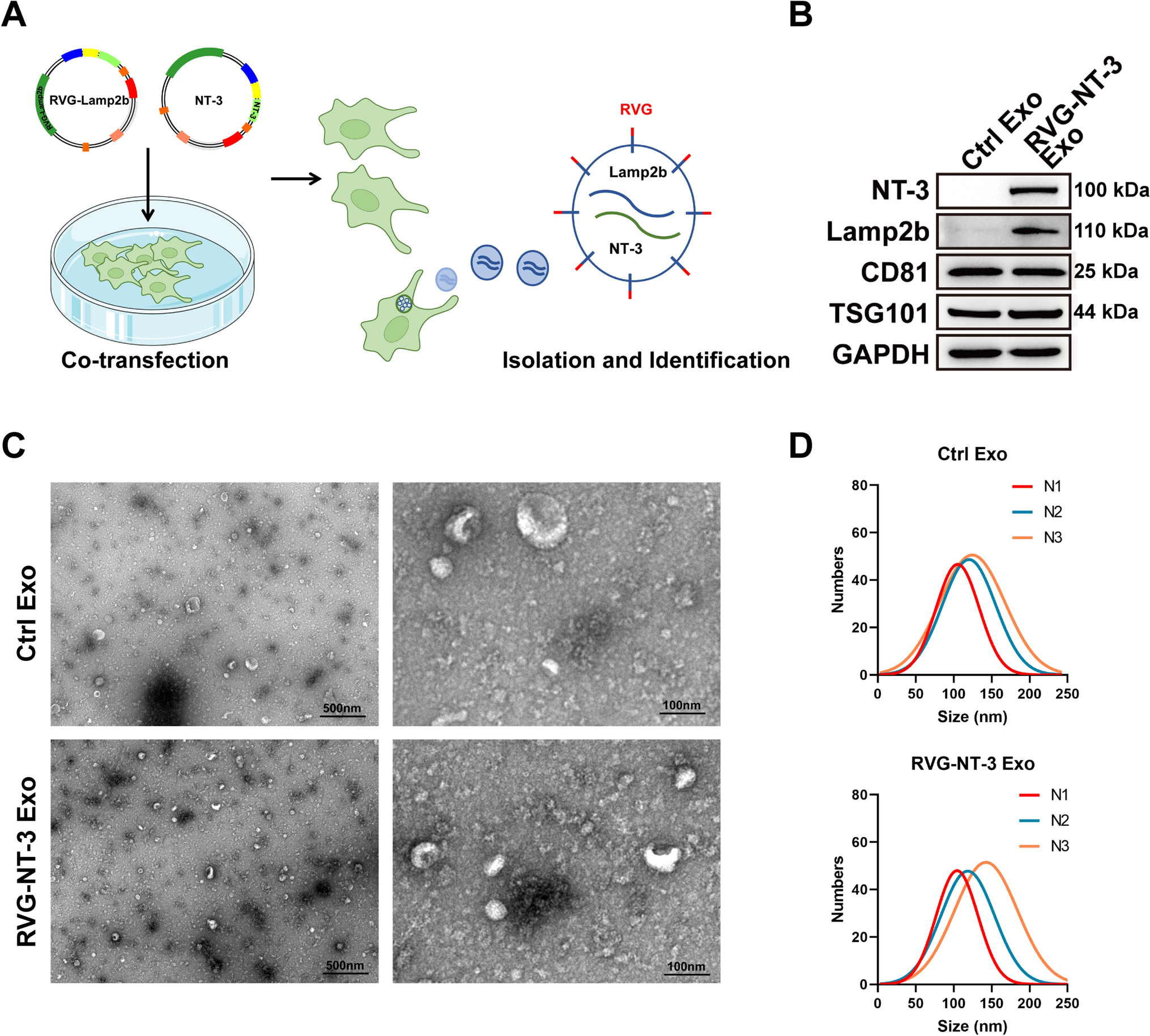
Bronchial epithelial cell culture (16HBE)
A human bronchial epithelial cell line immortalized with the origin-of-replication defective SV40 plasmid (pSVori-), 16HBE, were used in this study. 16HBE cells were maintained in a humidified atmosphere of 5% CO2 in air at 37 °C and were cultured as adherent monolayers. Eagle’s minimum essential medium (MEM), supplemented with 10% heat-inactivated (56 °C, 30 min) fetal bovine serum (FBS), 1% MEM (non-essential aminoacids), 2 mM L-glutamine and 0.5% gentamicin was used for culturing the cells.
Cell treatment
16HBE cells were grown to confluence for 24 h and then the culture medium was changed to a medium containing 1% FBS and without antibiotics. Thus, cells were exposed or not to different concentrations (10^6, 5*10^6, 10^7, 5*10^7, 10^8, 5*10^8, 10^9 colony-forming units (CFU)/ml) of a TB blend composed of Lactobacillus Casei, Lactobacillus Acidophilus, Lactobacillus Plantarum, Streptococcus Thermophilus. The experiments were repeated at least three independent times and each experiment was performed at least in duplicate.
Cell viability assay
The effect of TB on cell viability of 16HBE was evaluated as previously reported [20] by MTS assay using the CellTiter 96® Aqueous One Solution Cell Proliferation Assay (Promega, Madison, WI, USA), a colorimetric method capable of determining the number of viable cells by using 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethox-yphenyl)-2-(4-sulfopheyl) 2H-tetrazolium (MTS). 16HBE cells were seeded in 96-well plates and, after 24 h, were treated with the different concentration of TB above reported for 24 h. At the end of the treatment, 20 µL of CellTiter 96® AQueous One Solution reagent was added to each well and the plates were incubated for 20 min at 37 °C and 5% of CO2. The absorbance was measured by using a microplate reader at 490 nm (Microplate reader SPECTROstar-Nano, BMG-Labtech, Allmendgrün, Ortenberg). Results were expressed as percentage of viability compared with untreated cells (NT) (100% viability).
Staining of TB with SytoRed and internalization of TB
The bacterial suspension (at concentration of 10^9 CFU/ml) was incubated with SytoRed Fluorescent Nucleic Acid Stain (20 µM, Molecular Probes, Life Technologies, Thermo Fisher Scientific, MA, USA) for 30 min at room temperature in the dark. The stained suspension of bacteria was washed three times in PBS and then re-suspended in 1 ml of serum- and antibiotic-free medium. 16HBE cells were seeded in 96-well plates and, after 24 h, were incubated for 3 h with 100 µl of medium containing different concentrations of stained bacteria (10^6, 5*10^6, 10^7 CFU/ml). At the end of the incubation, the cells were washed with PBS and fixed with 100 µl 4% paraformaldehyde for 10 min at room temperature. After three washes, cells were stained with DAPI (Abcam, Cambridge, UK) and evaluated by the Operetta CLS system (PerkinElmer, MA, USA) in confocal mode at 63 × magnification.
Flow-cytometry
16HBE cells were seeded in 6-well plates and, after 24 h, were treated with 1 ml of medium containing different concentrations of SytoRed stained bacteria (10^6, 5*10^6, 10^7 CFU/ml) for 24 h. At the end of stimulation, cells were collected and incubated with a mouse FITC conjugated anti-human TLR2 (eBioscience, San Diego, CA). Negative controls were performed using mouse immunoglobulins negative control (Dako). E-Cadherin expression was also evaluated on 16HBE treated with different concentration of TB (10^6, 5*10^6, 10^7 CFU/ml) for 24 h. At the end of the treatment, cells were collected and incubated with E-cadherin antibody (sc-21791 Santa Cruz Biotechnology, Dallas, Texas, USA) followed by anti-mouse IgG FITC (Dako—Agilent Technologies, Santa Clara, CA, USA). Negative controls were set up using an isotype control antibody (BD PharMingen, Mountain View, CA). For both markers, cells were analyzed by flow-cytometry using a FACSCalibur (Becton Dickinson, Mountain View, CA) for TLR2 and CytoFLEX (Beckman Coulter, Brea, CA, USA) for E-Cadherin expression. Analysis was done on 10,000 acquired events for each sample. Cell debris and dead cells were excluded from the analysis. Data were expressed as percentage of positive cells.
Western blot analysis
The expression of E-cadherin in total proteins from cell lysates was assessed by western blot analysis as previously described [21]. Protein concentrations were evaluated using the Bradford assay (Biorad, Hercules, CA, USA). 30 μg of lysates were resolved on a 4–15% Mini-PROTEAN TGX Precast Protein Gels, then transferred to nitrocellulose membrane (Biorad, Hercules, CA, USA). The membranes were blocked with Odyssey Blocking Buffer (P/N 927–40,000) and probed with E-Cadherin antibody from Santa Cruz Biotechnology (sc-21791). The membranes were then incubated with IRDye® 800CW Donkey anti-Mouse (LI-COR Biosciences) and protein signal was then detected using the Odyssey® Classic Imaging System (LI-COR) and Image Studio™ Software (LI-COR). For loading control, membranes were re-tested with a mouse anti-β-Actin (Santa Cruz Biotechnology). Contrast and brightness were adjusted equally across entire images to best visualize protein bands.
Real‐time PCR
16HBE cells were seeded in 6-well plates and, after 24 h, were treated with different concentration of TB (10^6, 5*10^6, 10^7 CFU/ml) for 6 and 24 h. At the end of the treatment, the whole RNA was isolated using TRIzol Reagent (Life Technologies, Thermo Fisher Scientific, MA, USA) following the manufacturer’s instruction. 1 μg of RNA was reverse‐transcribed to cDNA, using iScript cDNA Synthesis kit (Biorad, CA, USA). TLR2, and E-cadherin (CDH-1) expression was evaluated by qRT‐PCR conducted by Step One Plus Real‐time PCR System (Applied Biosystems, Thermo Fisher Scientific, CA, USA) using specific FAM‐labeled probe and primers (prevalidated TaqMan Gene expression assay for TLR2, HS001872448_S1; for CDH-1, Hs001023895_m1; Applied Biosystems) as previously described [22, 23]. Gene expression was normalized to GAPDH (prevalidated TaqMan Gene expression assay for GAPDH, Hs03929097_g1) as endogenous control gene. The relative quantification of mRNA was obtained with the comparative Ct method (2^ − ΔΔCt) and was plotted as respective fold‐change. Untreated cells (NT) were used as reference sample.
ELISA
16HBE cells were seeded in 6-well plates and, after 24 h, were treated with different concentration of TB (10^6, 5*10^6, 10^7 CFU/ml) for 24 h. At the end of stimulation, the release of IL-6, IL-8, and TGF-β1 protein in cell supernatants was determined as previously reported [20] using human ELISA kit (R&D Systems, MN, USA) following manufacturer’s instructions.
Wound healing assay
16HBE were seeded in a 6-well plate and were cultured to confluence. Three circular wounds were prepared in each well using a 200-µl pipette tip. After washing with PBS 1X to eliminate debris, cells were allowed to recover for one hour and then incubated with different concentration of TB (10^6, 5*10^6, 10^7 CFU/ml) for 24 h. Digital images were acquired using a digital camera connected to an inverted phase-contrast light microscope at 0 h, 24 h and 48 h after wounding. The surface of the wound area was measured using the ImageJ software to assess remaining wound size and wound closure rates. The results were expressed as percentage of area reduction at time point 24 h (T24) compared to time point 0 h (T0) and at time point 48 h (T48) compared to time point 0 h (T0).
Statistical analysis
Data were expressed as mean ± SD and analyzed by analysis of variance (ANOVA). A p value < 0.05 was considered to be statistically significant. Correlation analyses between the cytokines IL-6, IL-8 and TGF-β1 were evaluated by calculating the parametric Pearson’s correlation coefficient r and a two-tailed p value. A p value of < 0.05 was considered statistically significant. These analyses and the correlation heatmap have been performed using R software (R, version 4.0.2; R Foundation for Statistical Computing: Vienna, Austria, 2020).







留言 (0)