
記住我



Myxofibrosarcoma (MFS) is a common adult soft tissue sarcoma that most frequently occurs in the elderly. The tumor shares overlapping genomic features with undifferentiated pleomorphic sarcoma (UPS), including widespread copy number alterations and often low tumor mutational burden (TMB, mutations/Mb) with mutations in TP53 and in genes associated with cell cycle checkpoints (RB1, CDKN2A).1–3 Moreover, methylation array studies have shown that MFS and most cases of UPS cluster similarly, implying that they constitute a morphologic spectrum of a single disease.4 Although MFS has distinct histopathologic features, no recurrent genetic alterations specific to MFS/UPS have been identified and, as a consequence, there are no effective targeted therapies.
The tyrosine kinase AXL, which drives cellular proliferation, is a single-pass transmembrane protein and a member of the Tyro3-AXL-MerTK family of receptors. AXL is activated in many cancers, with an important role in cell proliferation, motility, and the promotion of an immunosuppressive local microenvironment. In sarcomas specifically, AXL messenger RNA (mRNA) expression levels are significantly elevated in comparison with levels in other cancer types and in normal tissue.5 Among sarcomas, AXL mRNA expression is highest in MFS.5 Accordingly, a patient-derived xenograft study demonstrated that MFS showed the highest and most stable expression of AXL protein compared with other sarcoma types.6 Nevertheless, the pathogenic significance of these findings remains unknown. Characterization of AXL genomic alterations, either point mutations or copy number alterations (amplifications) in sarcomas, including MFS, has been limited.4,7 To assess the role of AXL abnormalities in MFS, we sought to characterize the genomics of AXL across sarcoma types. We identified 2 specific AXL mutations, as well as an unusually high frequency of AXL gene amplifications, as distinct genomic features of MFS.
METHODS Sample Selection and Comprehensive Genomic ProfilingOur archive of 463,546 tumor samples, including 19,879 sarcomas, 315 of which were MFS, each from a different patient, underwent comprehensive genomic profiling (CGP) in a Clinical Laboratory Improvement Amendments–certified, College of American Pathologists–accredited laboratory (Foundation Medicine Inc.). This cross-sectional study included patient samples that had been sent as part of routine care from medical care facilities across North America from January 2014 to March 2022 for identification of potentially targetable genetic alterations. Approval for this study, including a waiver of informed consent and a Health Insurance Portability and Accountability Act waiver of authorization, was obtained from the Western Institutional Review Board (Protocol No. 20152817). The present study was conducted according to the guidelines of the Declaration of Helsinki.
For CGP, sections were macrodissected to achieve >20% estimated percentage of tumor nuclei in each case, where percentage of tumor nuclei = 100 times the number of tumor cells divided by the total number of nucleated cells. Next, ≥60 ng DNA was extracted from 40 μm sections cut from tumor samples in formalin-fixed, paraffin-embedded tissue blocks. The samples were assayed by adaptor ligation hybrid capture, performed for all coding exons of 236 (v1), 315 (v2), or 405 (v3) cancer‐related genes plus select introns from 19 (v1), 28 (v2), or 31 (v3) genes frequently rearranged in cancer (Supplemental Table 1, Supplemental Digital Content 1, https://links.lww.com/PAS/B763).8,9 For samples with available RNA, targeted RNA‐sequencing was performed for rearrangement analysis in 265 genes.9 Sequencing of captured libraries was performed to a mean exon coverage depth of targeted regions of >500× using the Illumina HiSeq 4000 System. Sequences were analyzed for genomic alterations, including short variant alterations (base substitutions, insertions, and deletions), copy number alterations (focal amplifications and homozygous deletions), and select gene fusions or rearrangements.8,10,11 To maximize mutation detection accuracy (sensitivity and specificity) in impure clinical specimens, our CGP had previously been optimized and validated to detect base substitutions at a ≥5% mutant allele frequency, indels with a ≥10% mutant allele frequency with ≥99% accuracy, and fusions occurring within baited introns/exons with >99% sensitivity.8 Germline versus somatic status of pathogenic alterations was not delineated. Copy number analysis to detect homozygous deletions and gene-level amplifications at tetraploidy or greater was performed as previously described.8 TMB was determined on 0.8 to 1.1 Mbp of sequenced DNA.11 Microsatellite instability was determined on up to 114 loci.12
Analyses of Sarcoma Subtypes With Recurrent AXL MutationsFor the series of sarcoma cases sharing recurrent AXL mutations in a similar tumor subtype, we extracted clinicopathological data including patient age, sex, and tumor site from the accompanying pathology reports. Primary site data were not available for a subset of cases. Histopathology was reassessed on routine hematoxylin and eosin stain–stained slides of tissue sections submitted for genomic profiling by 2 board-certified pathologists (E.A.W. and E.A.M.).13
Differences among categorical variables were assessed using the Fisher exact test or the χ2 test with Yates continuity correction, depending on the number of patient cases. For comparisons of age and TMB between two groups, the nonparametric Mann-Whitney U test was used, to avoid assumptions of normality. The Bonferroni correction was applied for multiple comparisons, and a corrected 2‐tailed P value of <0.05 was considered statistically significant.
Review of Publicly Available Data SetThe Cancer Genome Atlas (TCGA) Network’s sarcoma genomic data set4 was interrogated for sarcomas carrying any recurrent AXL variant in a specific sarcoma lineage that we found in our own database. Histopathology of the single TCGA case identified in this manner was reviewed by 2 board-certified pathologists (E.A.W. and E.A.M.). Levels of AXL mRNA expression in this case and in the TCGA sarcoma data set overall were also examined.
AXL Complementary DNA Expression Vector Construction and Site-directed MutagenesisTo assess possible functional effects from a recurrent AXL gene mutation, we obtained the pLVX-TetOne-Puro-hAXL expression vector for wildtype (WT) AXL protein as a gift from Kenneth Pienta (Addgene Plasmid # 124797; http://n2t.net/addgene:124797; RRID:Addgene_124797). The AXL W451C mutation that we recurrently observed in human MFS samples (“Results”) was engineered into the pLVX-TetOne-Puro-hAXL construct by site-directed mutagenesis using the QuikChange II XL kit (Agilent) as directed by the manufacturer, to generate the pLVX-TetOne-Puro-hAXL(c.G1353C, p.W451C) expression vector. Primer sequences used for the mutagenesis reactions were as follows, with the mutated nucleotide underlined and the mutated sequence in the expression vector subsequently verified by Sanger sequencing:
AXL p.W451C Fwd: 5′-gccttctcgtggccctggtgctatgtactgctaggagcag-3′
AXL p.W451C Rev: 5′-ctgctcctagcagtacatagcaccagggccacgagaaggc-3′
Lentiviral Production and InfectionThe pLVX-TetOne-Puro-hAXL WT and the mutant hAXL(c.G1353C, p.W451C) expression vectors were separately cotransfected into HEK-293T cells with the lentiviral packaging carrier plasmid pLP VSV-G (Nova Lifetech, Cat# PVT2326) and the lentiviral packaging plasmid psPAX2 (a gift from Didier Trono, Addgene plasmid # 12260; http://n2t.net/addgene:12260; RRID:Addgene_12260) using TransIT-Lenti Transfection Reagent (Mirus, Cat# MIR 6604) following the manufacturer’s recommended protocol. Virus-containing medium was harvested 48 hours after transfection, purified by passage through a 0.45 μm pore-size filter, and used to transduce recipient HEK-293T cells in the presence of 8 μg polybrene/mL (Sigma-Aldrich, Cat# TR-1003). The transduced cells were selected by 2 μg puromycin/mL. Induction of expression of WT and W451C-mutant AXL protein was achieved with 0.1 μg doxycycline/mL, applied 96 hours before harvesting cellular protein for further analyses. Negative control HEK-293T cells were transduced and selected, but received buffer instead of doxycycline.
ImmunoblotsTotal protein was extracted from transduced HEK-293T cells by adding lysis buffer (1% Triton X-100, 0.5% NP-40, 150 mM NaCl, 50 mM Tris-HCl, pH 7.4, 10% glycerol) supplemented with inhibitors of proteases (Roche, Cat# 11836170001) and phosphatases (Roche, Cat# 4906837001). Protein concentration was determined by using the Detergent-Compatible Protein Assay (Bio-Rad, Cat# 500-0116). Cellular proteins were resolved by Sodium dodecyl-sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and then immunoblotted following standard biochemical techniques, under nonreducing (6X sample buffer: 375 mM Tris-HCl, pH 6.8, 9% SDS, 0.03% bromophenol blue, 50% glycerol) and reducing (4X sample buffer: 200 mM Tris-HCl, pH 6.8, 8% SDS, 0.4% bromophenol blue, 40% glycerol, 5% β-mercaptoethanol) conditions.
Primary antibodies detected phospho-AXL at Tyr779 (pY779-AXL, Invitrogen, Cat# MA5-24334), total AXL (t-AXL, Cell Signaling, Cat# 8661), phospho-ERK1/2 at Tyr204 (pY204-ERK, Cell Signaling, Cat# 4377S),14 total ERK1/2 (t-ERK, Cell Signaling, Cat# 9102), α-tubulin (Santa Cruz Biotechnology, Cat# SC-23948), and β-actin (Sigma-Aldrich, Cat# A5441). The last 2 targets were used as loading controls. All primary antibodies were used at 1:1000 dilutions, except antibodies to detect the loading controls, which were used at 1:5000 dilutions. The signal was developed by using WesternBright™ ECL (Cat#K-12045) from Advansta, USA. The band intensity was quantified by ImageJ 1.54d software. The significance in the protein level was calculated by Prism 9 using unpaired t-test analysis.
RESULTSThe single nucleotide variant (SNV) status of the AXL gene (NM_021913) was reviewed in each of the 19,879 unique sarcomas in our own database. Two hundred thirty-six SNVs of AXL were identified in 228 sarcomas, listed in Supplemental Table 2 (Supplemental Digital Content 2, https://links.lww.com/PAS/B764). Only 29 of these variants, however, were recurrent (displayed in Supplemental Table 3, Supplemental Digital Content 3, https://links.lww.com/PAS/B765). The sole AXL mutations for which there were multiple cases of the same sarcoma subtype were W451C and W450C. Each of the remaining 27 recurrent SNVs that we identified did not show recurrence in a single sarcoma subtype (Supplemental Table 3, Supplemental Digital Content 3, https://links.lww.com/PAS/B765). Detailed clinicopathologic and genomic features of the 8 AXL W451C-mutant and 2 AXL W450C-mutant sarcomas are shown in Table 1. A single patient case had both primary tumor and lymph node metastasis sequenced sites (case #3), each of which harbored an AXL W451C mutation. Of note, the variant allele frequency for the AXL alteration in the 11 samples was high (median: 31%, Supplemental Table 2, Supplemental Digital Content 2, https://links.lww.com/PAS/B764). No AXL W451C mutations were identified in any of the 443,667 non-sarcoma tumors.
TABLE 1 - Detailed Clinicopathologic and Molecular Features of All 9 AXL W451C-mutant (cases #1-9) and 2 AXL W450C-mutant (Cases #10, 11) Sarcoma Patient case Age (y)/sex Primary tumor site Anatomic location of the sequenced tumor Primary tumor size (cm) Diagnosis Grade (FNCLCC) Mitoses/10 HPF Necrosis Immunohistochemistry TMB (mut/Mb) Genomic alterations (in addition to AXL mutations) Disease and/or survival status 1 74/M Left thigh Left thigh At least 14.9 MFS 3 8 Present (<50%) CD34: focally positive*TCGA case.
CBD indicates cannot be determined; FNCLCC, Fédération Nationale des Centres de Lutte Contre le Cancer (National Federation of Centers for the Fight Against Cancer); IHC, immunohistochemistry; HPF, high power field.
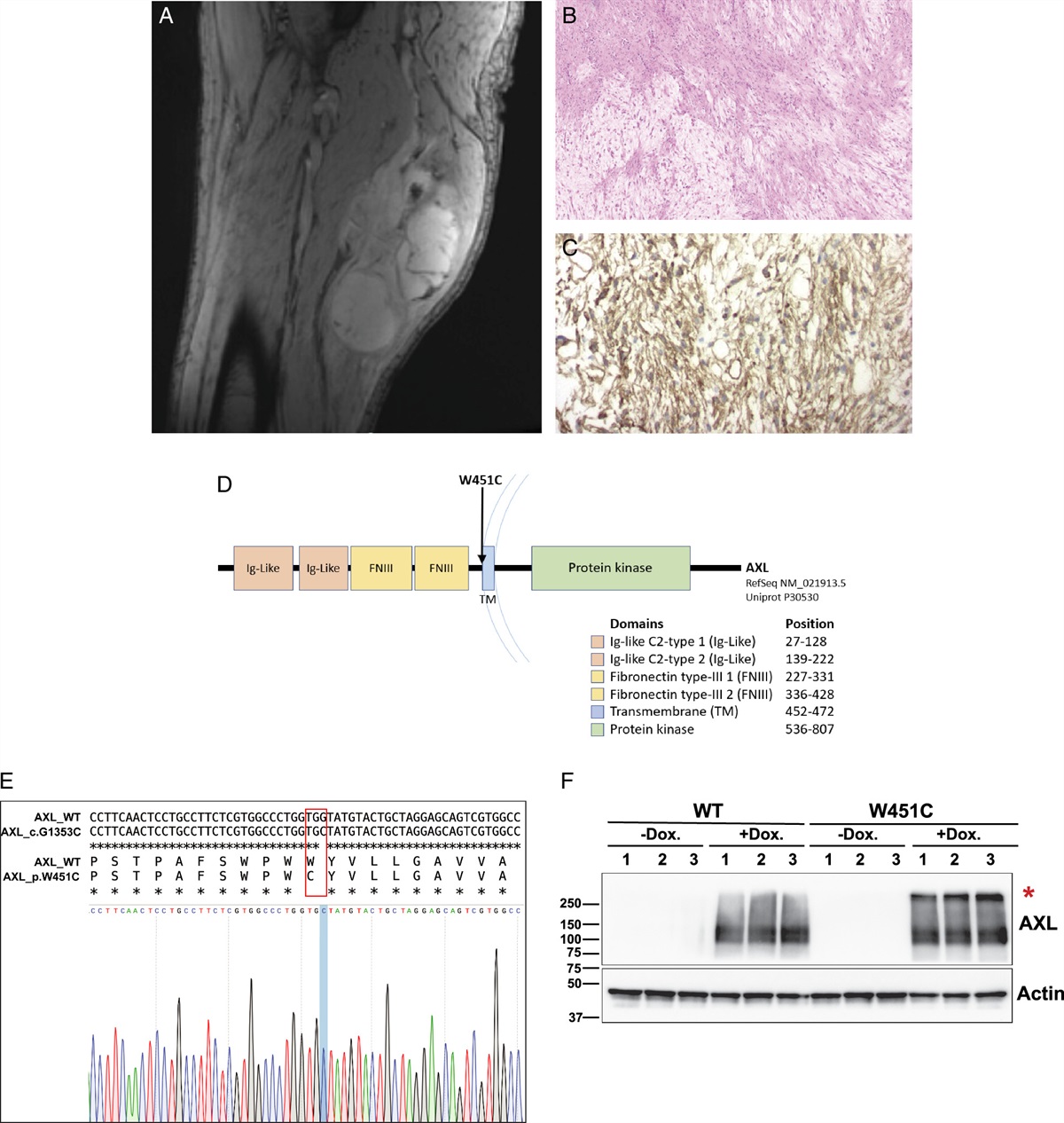
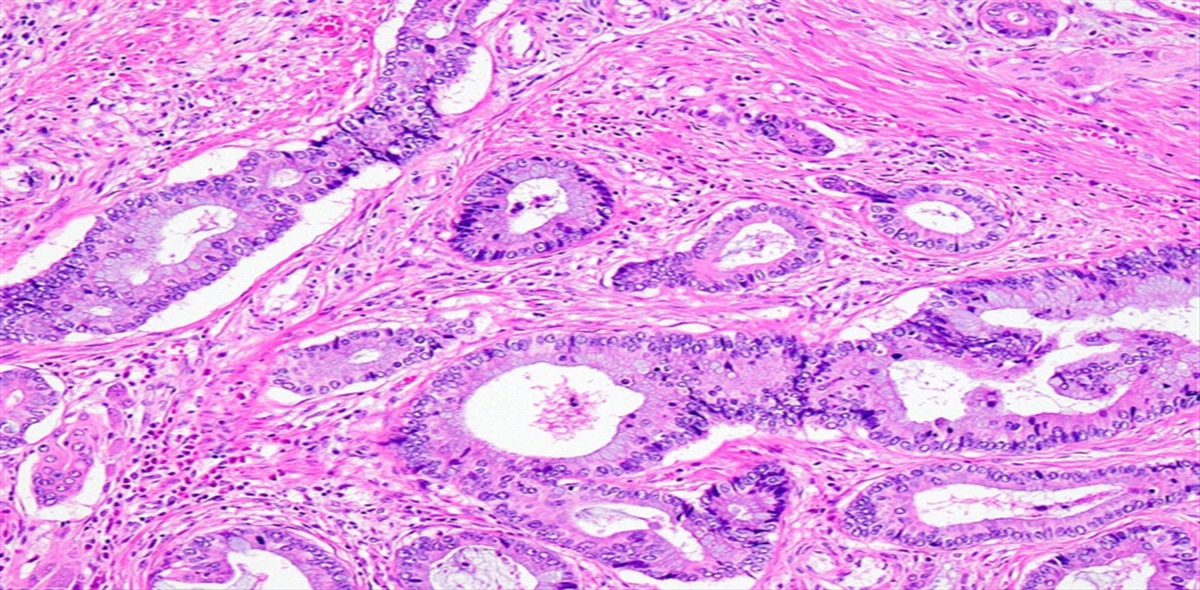
Clinical imaging, histopathology, and immunohistochemistry of case #1 are shown (Fig. 1A–C), along with a diagram of the protein structure of AXL and the W451C mutation site, which is located at the base of the extracellular domain of the protein product (Fig. 1D). Characteristic histopathologic features of additional AXL W451C-mutant cases are showed in Supplemental Figure 1 (Supplemental Digital Content 4, https://links.lww.com/PAS/B766).
 FIGURE 1:
FIGURE 1: Index patient with AXL W451C-mutant MFS. A, T2-weighted MRI at initial presentation of the left thigh demonstrates a large multiseptated mass with internal T2 hyperintensity suggestive of myxoid components (case #1). Histopathologic examination showed prominent myxoid material (B); hematoxylin and eosin stain) and there is a focal strong expression of CD34 on immunohistochemical staining (C). On genomic profiling, the MFS harbored an AXL W451C mutation, corresponding to the base of the extracellular domain of the protein product, as depicted in the diagram, in which the blue arc represents the plasma membrane (D). Sequencing also showed a TP53 p.R249G mutation and homozygous loss of CDKN2A/B. An AXL W451C expression vector was developed and verified by Sanger sequencing (E). HEK-293T cells were transduced with either WT AXL cDNA or AXL W451C, and a nonreducing Western blot of protein lysates extracted from these cells revealed that AXL W451C, and not AXL-WT, resulted in dimerization (red asterisk; (F). cDNA indicates complementary DNA; MRI, magnetic resonance imaging; TM, transmembrane.
The AXL W451C-mutant and W450C-mutant cases showed similar clinical features and similar concurrent molecular alterations as in AXL-WT MFS, which we defined as MFS without AXL W451C or W450C mutations and without AXL gene amplification (Table 2).
TABLE 2 - Comparative Demographics and Percentage Frequency of Genomic Alterations Stratified by AXL Status, With P Values. AXL W451C and W450C sarcoma (SNV) P (SNV vs. WT) AXL-amplified MFS P (amplified vs WT) AXL-WT MFS No. cases 10 — 8 — 301 % male 80 (8/10) 0.33 75 (6/8) 0.48 60 (180/301) Median age (y); range 72 (44-81) 0.42 72 (21-84) 0.27 66 (16-89+) TMB (mut/Mb); Q1-Q3 3.9 (2.5-4.1) 0.0051 3.2 (2.0-4.4) 0.12 2.4 (1.6-3.5) % MSI high 0 (0/10) 1 13 (1/8) 0.051 0.3 (1/301) TP53 20 (2/10) 0.023 63 (5/8) 1 58 (174/301) CDKN2A 80 (8/10) 0.0026 50 (4/8) 0.27 31 (94/301) RB1 20 (2/10) 0.73 0 (0/8) 0.11 29 (88/301) ATRX 10 (1/10) 0.70 0 (0/8) 0.36 19 (56/301) NF1 0 (0/10) 0.22 25 (2/8) 0.64 18 (55/301) PTEN 10 (1/10) 0.51 0 (0/8) 1 7 (20/301) KRAS 20 (2/10) 0.052 0 (0/8) 1 3 (10/301)For percentage values, the number of positive cases over the number of evaluated cases is included in parentheses.
The Bonferroni correction for 22 simultaneous comparisons was applied, with the cutoff for significance at P = 0.05/22 = 0.002.
MSI indicates microsatellite instability.
To assess the role of additional AXL gene abnormalities in MFS, we also reviewed AXL copy number alterations. Sarcoma cases with gene-level amplifications at tetraploidy or greater are shown in Supplemental Table 4 (Supplemental Digital Content 5, https://links.lww.com/PAS/B767). Cases diagnosed as MFS showed more frequent AXL copy number gain compared with other common sarcoma categories and with sarcomas overall (Supplemental Table 5, Supplemental Digital Content 6, https://links.lww.com/PAS/B768). In particular, 2.5% of MFS showed tetraploid or greater AXL amplification compared with only 0.5% of non-MFS sarcoma (8/315 vs 96/19564, P < 0.0001, χ2 test with Yates correction). MFS with AXL amplification showed similar clinical and molecular features to AXL-WT MFS (Table 2). We also reviewed sarcomas with AXL gene rearrangements (Supplemental Table 6, Supplemental Digital Content 7, https://links.lww.com/PAS/B769), but no recurrent partners were identified and no recurrent breakpoints that preserved the AXL kinase domain were seen within any single sarcoma category.
The recurrence of the 2 AXL point mutations in our sarcoma data set (n = 10/19,879) prompted us to interrogate an additional database—namely, the sarcoma genomic data set of the TCGA Network. A single AXL W451C-mutant sarcoma was identified (one of 206 sarcomas). The case (TCGA-DX-A8BX-01A) occurred in a 52-year-old man with UPS and harbored a concurrent point mutation in PIK3CA (Table 1). Available data on RNA expression showed that the AXL mRNA level, in this case, was not conspicuously elevated, falling within the second quartile of sarcoma overall (6.48 transcripts/million; Q2 for sarcomas spans 5.8 to 6.7). Molecular analysis revealed that the tumor clustered into the MFS/UPS group across multiple platforms (mRNA expression, DNA methylation, and PARADIGM profile; data not shown).4
Based on our discovery of AXL mutations that encode a new cysteine at the base of the extracellular domain (Fig. 1D, E), we hypothesized that the novel AXL W451C mutation might allow the creation of an abnormal disulfide bond between pairs of mutant proteins expressing ectopic cysteine residues, resulting in unregulated dimerization of the mutant AXL protein, hence autophosphorylation and activation of downstream signaling. To test our hypothesis, we transduced HEK-293T cells with lentiviral expression vectors for either the full-length WT AXL protein or the W451C AXL mutant (verified by Sanger sequencing, Fig. 1E). Cells were treated without (negative control) or with doxycycline to activate the expression vectors.
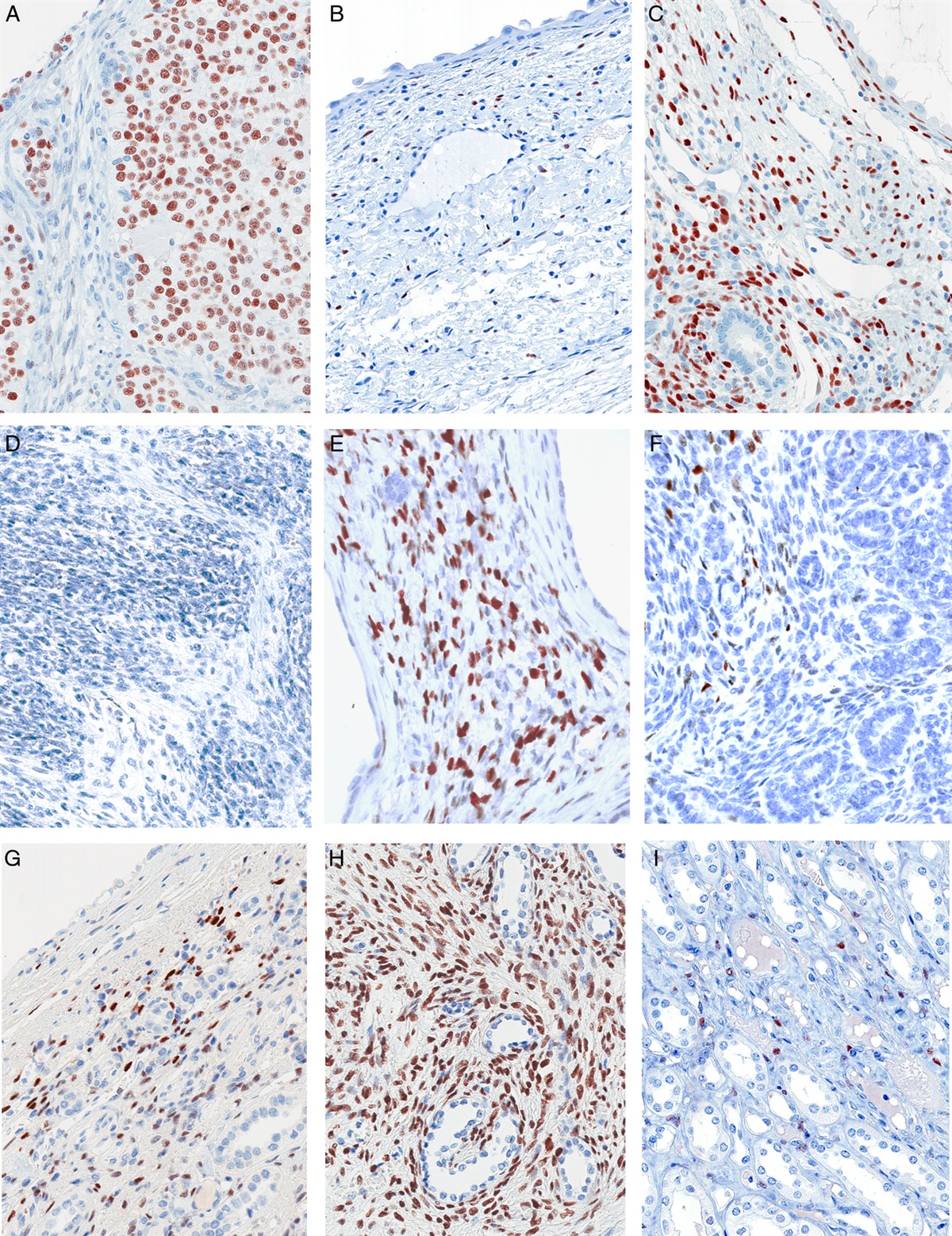
Protein lysates extracted from these cells were evaluated by SDS-PAGE under nonreducing conditions, followed by immunoblotting with an antibody against total AXL protein (meaning phosphorylated and nonphosphorylated forms). Figure 1F shows that after induction of the W451C AXL mutant, it spontaneously dimerized, whereas the WT AXL protein did not (Fig. 1F).
To assess functional effects, protein extracts from these cells were evaluated by SDS-PAGE under nonreducing conditions, followed by immunoblotting with an antibody specific for AXL protein that has been phosphorylated at its activation site, Tyr779 (pY779-AXL). Figure 2A shows that the dimerized form of the W451C AXL mutant was phosphorylated at this key site, whereas the small amount of higher-molecular-weight WT AXL exhibited essentially no Tyr779 phosphorylation. Monomers of neither the mutant nor the WT form showed any substantial Tyr779 phosphorylation (Fig. 2A).
 FIGURE 2:
FIGURE 2: Western blots of protein lysates extracted from HEK-293T cells transduced with WT and mutant AXL isoforms. A nonreducing western blot evaluated with phospho-AXL antibody revealed phosphorylation of the dimerized form (red asterisk, (A), with a significant increase in phosphorylation as compared with WT (A). By contrast, in a reducing western blot, there was minimal evidence of dimerization (red asterisk; (B). For the monomer form seen in this reducing condition, phosphorylation of AXL is increased in AXL W451C as compared with AXL-WT (B). Expression of AXL W451C-mutant isoforms leads to increased ERK1 (p44) and ERK2 (p42) phosphorylation, as compared with AXL-WT (C) (*p<0.05, **p<0.01).
In our immunoblots prepared under reducing conditions that break disulfide bonds between cysteine residues, essentially no AXL dimers remain (Fig. 2B). Accordingly, under reducing conditions, all of the pY779-AXL signal appears at the molecular weight of AXL monomers, and phosphorylation of AXL is still increased in the AXL W451C-mutant over AXL-WT (Fig. 2B).
To assess downstream signaling, we found that expression of the W451C AXL mutant significantly increased ERK phosphorylation at its Tyr204 site (pY204-ERK), compared with the effect from expression of the AXL-WT (Fig. 2C).
DISCUSSIONIn the current study, we sought to assess a possible role for AXL abnormalities in MFS through a search for AXL genomic alterations across large data sets of sarcomas. We found 2 specific novel recurrent AXL mutations, W451C and W450C, as well as unusually high levels of AXL gene amplification, as distinct genomic features of MFS. Moreover, the TCGA data set indicates high levels of AXL mRNA expression in MFS, even in comparison with other sarcomas.5 Our functional studies revealed that the AXL W451C mutation causes abnormal dimerization of the protein, autophosphorylation, and ligand-independent activation of downstream signaling (gain-of-function). Taken together, our findings implicate activating aberrations in AXL as a distinct feature of MFS and might aid in diagnosis as well as selection of available targeted therapies.
AXL is a receptor tyrosine kinase that, upon binding its ligand, growth arrest-specific gene-6, dimerizes and thereby activates key signaling pathways. Inhibitory antibody conjugates and small-molecule inhibitors have been developed that target the AXL protein and inhibit its signaling.6,15–17
MFS is a sarcoma with distinct histopathologic features18 but only nonspecific genomic findings identified previously, including typically low TMB with alterations in TP53 and genes encoding proteins in the cell cycle.1,2 Prior methylation array studies have shown that MFS and most UPS cluster together, supporting that they represent a histopathologic spectrum of a single disease.4 MFS shows the highest AXL mRNA expression level in the TCGA data set compared with normal tissues, other cancer types, and even other sarcoma types.5 Of note, the single AXL W451C-mutant case in the TCGA did not show markedly increased AXL mRNA levels.4 This finding is consistent with our functional studies showing that the W451C mutation enhances AXL signaling even at relatively unremarkable absolute levels of expression.
Amongst AXL-WT cases of MFS, which we defined as MFS without AXL W451C or W450C mutations and without AXL gene amplification, we confirmed prior work on the TCGA data set indicating high levels of AXL mRNA.5 This finding raises the possibility of critical epigenetic processes in AXL-WT MFS that upregulate AXL expression or signaling. A potentially analogous process is the modulation of MED12 in leiomyosarcoma, in which MED12 point mutations are rare events that disrupt MED12 function, while the majority of leiomyosarcoma show epigenetic downregulation of MED12.19 In MFS, the AXL W451C point mutation is a rare, but specific, alteration that appears to characterize a subset of the disease, while more generally, AXL mRNA expression is markedly elevated in a majority of MFS cases. Overall, our findings, in combination with prior work,5,6 suggest that AXL activation is a distinguishing and possibly defining feature of MFS/UPS and could become a specific diagnostic tool and therapeutic target. We propose that these rare tryptophan-to-cysteine mutations provide an important clue to the pathogenesis of MFS.
Both W451C and W450C alterations are tryptophan-to-cysteine, resulting in a new, potentially reactive thiol at the base of the AXL extracellular domain. Our functional studies showed that the W451C substitution allows the formation of disulfide bridges that cause ligand-independent AXL dimerization and activation. This mechanism of activation has been described for other receptor tyrosine kinases. For example, ligand-independent activation of ERBB2 has also been described as mediated by abnormal disulfide bond-induced dimerization resulting from cysteine substitutions in the juxtamembrane domain.20 Similarly, for DDR1, site-directed cysteine substitutions in the extracellular juxtamembrane region cause high-efficiency formation of covalent dimers, resulting in autophosphorylation.21
Limitations of this study include its retrospective nature and enrichment for tumors from patients with aggressive disease, including many neoplasms metastatic to distant sites. The submission of specimens for sequencing was presumably driven by late-stage disease. In addition, methylation array studies were performed only on the TCGA case in this study, which is particularly relevant for case #7, which was diagnosed as a low-grade sarcoma, not otherwise specified as MFS or UPS.
Additional studies are needed to identify if targeting AXL will benefit patients with MFS/UPS. A xenograft model showed that, among sarcomas, the only xenograft mice with significantly improved survival upon treatment with an inhibitory anti-AXL antibody conjugate were MFS/UPS cases.6 Several AXL-targeted therapeutics are in early clinical trials. Based on previous work15,16 and our current findings, those therapeutics could be explored with a particular focus on MFS.
Future studies could evaluate other diagnostic modalities, such as AXL testing through immunohistochemical surrogates. Regarding the AXL protein itself, we do not yet know whether the high levels of AXL mRNA in most MFS/UPS or high levels of AXL gene amplifications translate into high levels of AXL protein that could be detected clinically by immunohistochemistry. Additional studies are also needed to identify possible epigenetic events that could activate AXL expression and activity in MFS as possible novel therapeutic targets. In summary, our CGP of MFS has revealed recurrent, specific, activating alterations in AXL that may provide insights into MFS biology and potentially inform diagnostic and therapeutic options, including AXL-targeted agents.
REFERENCES 1. Ogura K, Hosoda F, Arai Y, et al. Integrated genetic and epigenet
留言 (0)