
記住我








Various neurodevelopmental atypicalities are overrepresented in males, and autism spectrum conditions (ASC), which have been the targets of primary attention, are approximately three times more prevalent in males than females (1) [however, some current arguments maintain equal prevalence of ASC across sexes (2)]. There are also global sex differences in professional interest and cognitive strength (3, 4). Classically, Geshwind et al. assumed that high prenatal androgen levels cause the suppression of “dominant” left cerebral cortex language field development, leading to learning disorders, left-handedness, and susceptibility to immune diseases and migraines (5, 6). In a more recent version, Baron-Cohen’s Extreme Male Brain (EMB) theory posits that systemizing-empathizing dimensions of cognitive preference explain ASC tendencies in parallel with male-female brain differences (7–9). There is a global sex difference in the 2nd to 4th digit ratio, and a smaller (< 0) ratio is regarded to reflect a stronger androgen effect in utero. The finger digit ratio is easy to measure and came to be the most popular putative biomarker of prenatal androgen effects, instead of handedness (10).
However, these premises are rebutted by many accounts that failed to find an association between various androgen measures and ASC symptoms (11), especially in ASC males (12). Brain differences between ASC versus typically developed (TD) in women somewhat overlap with male-female sex differences among TD, but they have little overlap with ASC-TD differences within men (13). The diagnosis of ASC does not imply a uniform tendency, which covers a multifaceted spectrum (14), questioning the plausibility of research strategies that investigate mechanisms in parallel with typical gender differences.
Therefore, we focused on multiple chromosomal/genetic syndromes associated with sex steroid deficiencies and the known risks of ASC. About 5–20% of ASC are considered syndromic (15), and many also show hypogonadism. Characteristically, 30–50% of individuals with Klinefelter syndrome (KS), one of the most prevalent syndromes with chromosomal aneuploidy, have ASC, in contrast to the prevalence rate of 1% in the general population (16). In other syndromes with hypogonadism, 11–80% of Prader-Willi syndromes (PWS) are associated with ASC (15). Fragile X and Down syndromes also show a high prevalence of ASC along with other neurodevelopmental disorder (NDD) symptoms and psychiatric status. Hundreds of genes related to developmental or psychiatric status overlap across different diagnostic categories (17, 18). The diagnostic criteria category is not as typological as previously believed. The viewpoint of capturing status as a spectrum covering diverse phenotypes has become predominant (14). When investigating the effect of sex steroids on cognitive function, it would be more promising to focus on syndromes with known physiological factors and common endophenotypes instead of being bound by the Diagnostic Statistical Manual criteria (19).
Another drawback of the EMB theory in explaining individual differences in strength and weakness across cognitive domains, in parallel with general sex differences, is that the prevalence of non-heterosexuals among the ASC population is nearly twice that of TD (20–22). At the same time, the prevalence of ASC in male and female transgender people is approximately 7–10 times than in the general population (23, 24). Furthermore, mediation analysis suggests that the non-heterosexuality of individuals with ASD is mediated by a higher incidence of gender dysphoria (20). Among KS individuals, some of whom experience hypogonadism in utero, and self-perceived gender nonconformity tends to co-emerge with NDDs (25). These pieces of evidence suggest a common underlying mechanism between NDD risk and atypical gender identity development.
In this paper, we have collected evidence that non-correlative biopharmacological studies support that hypoestrogenism is a more robust predictor of NDDs, and a cluster of outstanding abilities. The apparent correlation between hyperandrogenism and such cognitive traits can be explained as the result of negative feedback and a pseudo-relationship (26, 27) (see section 2.1.3). This paper connects evidence from atypical sexual development with the neurodevelopmental theory of ASC, which is based on the free energy principle by Karl Friston. Oxytocin plays an important role in early insular development by helping neural predictive signals synchronized with external and internal stimuli (28), leading to a sense of self-integration (29)(Figure 1). The insula is a central part of integrating external and internal sensations into self-image (28) and switches between the self-referential Default Mode and Central Executive Networks in the neural process of cognition (30). Steroids and oxytocin bidirectionally and synergistically affect early neural development and function in adults, and disruption of oxytocin activity has been reported in PWS, which shows hypogonadism and a strong tendency toward ASC (31, 32). In the rat brain, estrogen receptor beta (ERβ) is distributed over most cerebral cortex areas, mainly in layer V. Estrogen receptor beta is abundant in the primary motor and somatosensory fields, and has a lower density in the insula, followed by the cingulate cortex (33). The cingulate cortex works together with the insula and connects incoming sensation with emotional recognition and control. A recent hypothesis regarding schizophrenia suggested that insufficient estrogen and oxytocin levels are upstream etiologies. Moderate increases in these hormones ameliorate the positive and negative symptoms of schizophrenia by attenuating impairments in prepulse inhibition, resulting in the facilitation of emotion recognition and social interaction (34).
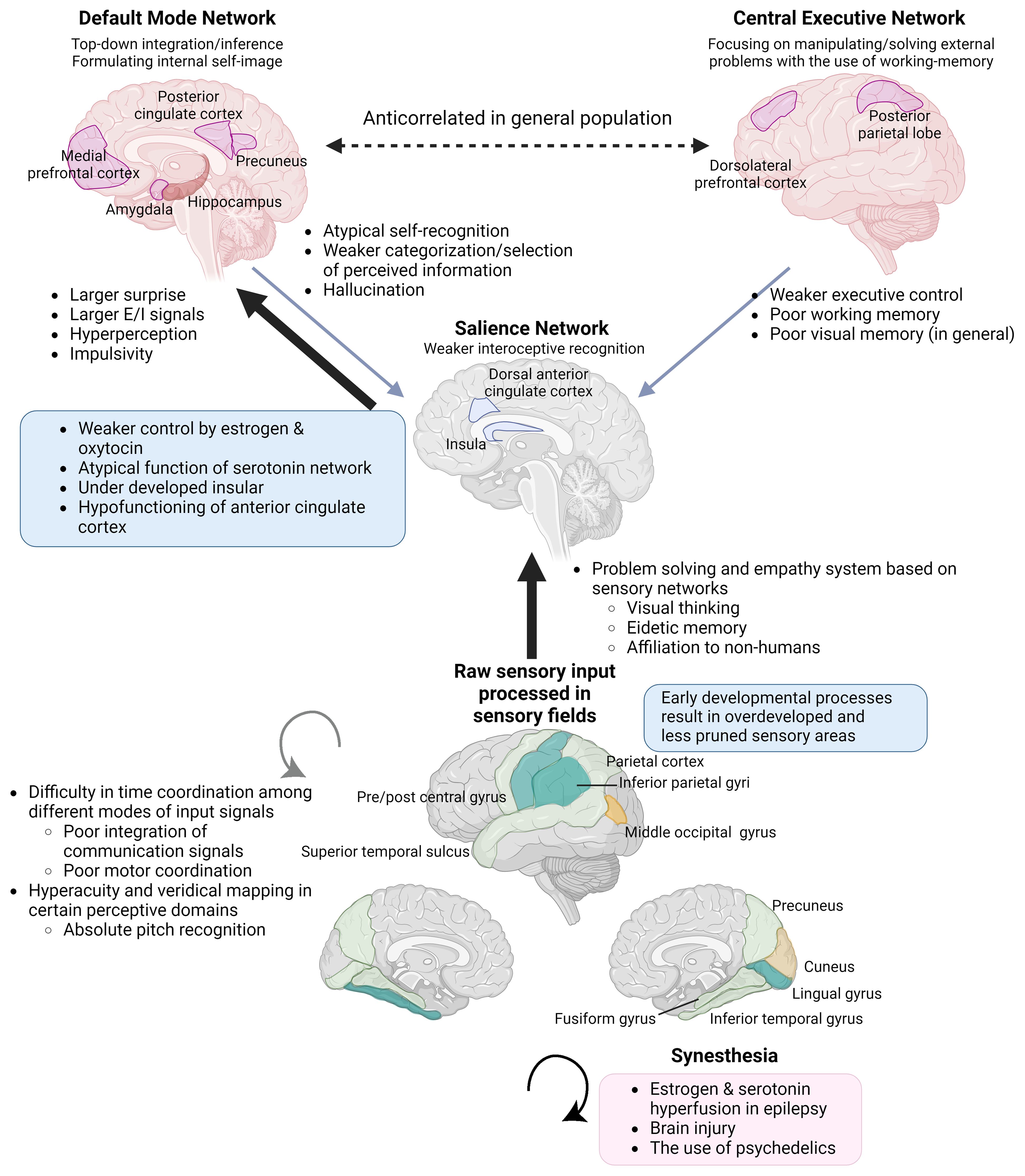
Figure 1 Integration of information processing theories and endocrinological characteristics underpinning endophenotypes of ASC, KS, and savant/gifted. The Enhanced Perceptual Functioning model and the neurodevelopmental theory based on the free energy principle, in combination with an understanding of the functions of brain networks and the interoceptive feedback system, can explain endophenotypes commonly observed across individuals with NDDs and savants/gifted individuals. The significance of each aspect varies across syndromes and subtypes within them. Brain images at the bottom. Yellow: the surface expanded areas in infants with high ASC risks. Light green: the thicker cortical areas in male to female transgenders. Green: the overdeveloped parts common to the two groups (Section 3.1.2). Individuals with KS also show similar overdevelopment patterns (Section 3.2.3). E/I, excitation/inhibition. (Created with BioRender.com).
The brains of individuals with KS (16), ASC (35, 36), and schizophrenia (37) commonly show the under-development or function of the insula. Additionally, KS (16), ASC (38), and also male-to-female transgender individuals (39) share commonalities in the overdevelopment of primary sensory areas (Figure 1). Underdevelopment of the insula can lead to overfocusing on raw sensory input processed in sensory areas and an inability to abstract perceptive stimuli, resulting in shortcomings in emotional regulation, motor coordination, and the processing of communication signals, including language (28, 40, 41). This paradigm also helps to focus on the co-occurrence of endophenotypes across different diagnostic categories, such as ASC, attention-deficit hyperactivity disorder (ADHD) (42), schizophrenia, dyslexia, along with atypical self-integration, and mystical thinking, which might become the source of wild imagination and creative inspiration (43).
2 Androgyny hypothesis of ASC and underlying mechanism of cognitive phenotype2.1 Estrogen deficiency instead of androgen excess hypothesis of ASC2.1.1 Syndromes with hypogonadism known as risk factors for ASC and other developmental uniquenessThe most notable drawback of the EMB is the high prevalence of ASC in several hypoandrogenic populations (44, 45). Klinefelter syndrome (XXY and its variations with >2 Xs) is the most prevalent sex chromosome aneuploidy (SCA), with a high end of prevalence rate of ≈1:500 in males (46). The lifetime diagnosis rate of KS is low, for example UK, 36% (47) and 23% (48); Australia, 50% (49); and Denmark, 25% (50), and approximately one-tenth of them are diagnosed prenatally (47). While typically known for tall stature, eunuchoid physique, learning difficulties, and impulsivity, the emergent phenotypes of KS are diverse. In a population who had been diagnosed irrespective of symptoms, the prevalence of gynecomastia and delay in school achievement did not differ from that in a control population (51). Common and robust characteristics include microorchidism, hypergonadotropic hypoandrogenism, and infertility (16). Hypoandrogenism is observed in > 75% of the diagnosed cases (16).
Among individuals diagnosed with KS, 30–50% are affected by ASC (16), and 63% are diagnosed with ADHD (52). The incidence of schizophrenia has been reported to be high; however, this observation was based on a small clinical sample (53). Psychosocial atypicality is generally considered to emerge together with hypogonadism features (54), whereas there have been case reports of ASC manifestation together with typical androgen production (55). A greater number of X-chromosomes leads to more pronounced physiological and cognitive characteristics (56, 57). In KS, ASC characteristics are mild, and restrictions of interest do not emerge (58). Verbal intelligence quotient (IQ) is typically in the low-normal range, whereas performance IQ measuring visuospatial ability is not impaired (59) and sometimes reaches a gifted range (>130) (60). Boys with KS show relative strength in arithmetic (59), although some report weakness in arithmetic problem solving. Executive function is affected by diminished cognitive flexibility and reduced working memory (53, 61). Testosterone supplementation improves language skills and concentration (53) (Table 1), but not visuospatial ability, which is generally considered to be promoted by androgens. Hypoandrogenism develops after puberty; however, speech and motor delays appear before that. Androgen secretion in the peripheral and central neural systems prenatally (165) and infantile mini-puberty (166) tends to be low, implying its influence on early developmental stages. Prenatal androgen production rate is diverse, as manifested in the micropenis manifestation in 10–25% of diagnosed cases (16). Hypogonadotropic hypogonadism cases are included in such a phenotype, and the percentage is as low as two in 160 KS individuals (167). Decrease in bone mineral density is observed in <40% individuals, indicating lower estrogen function (16).
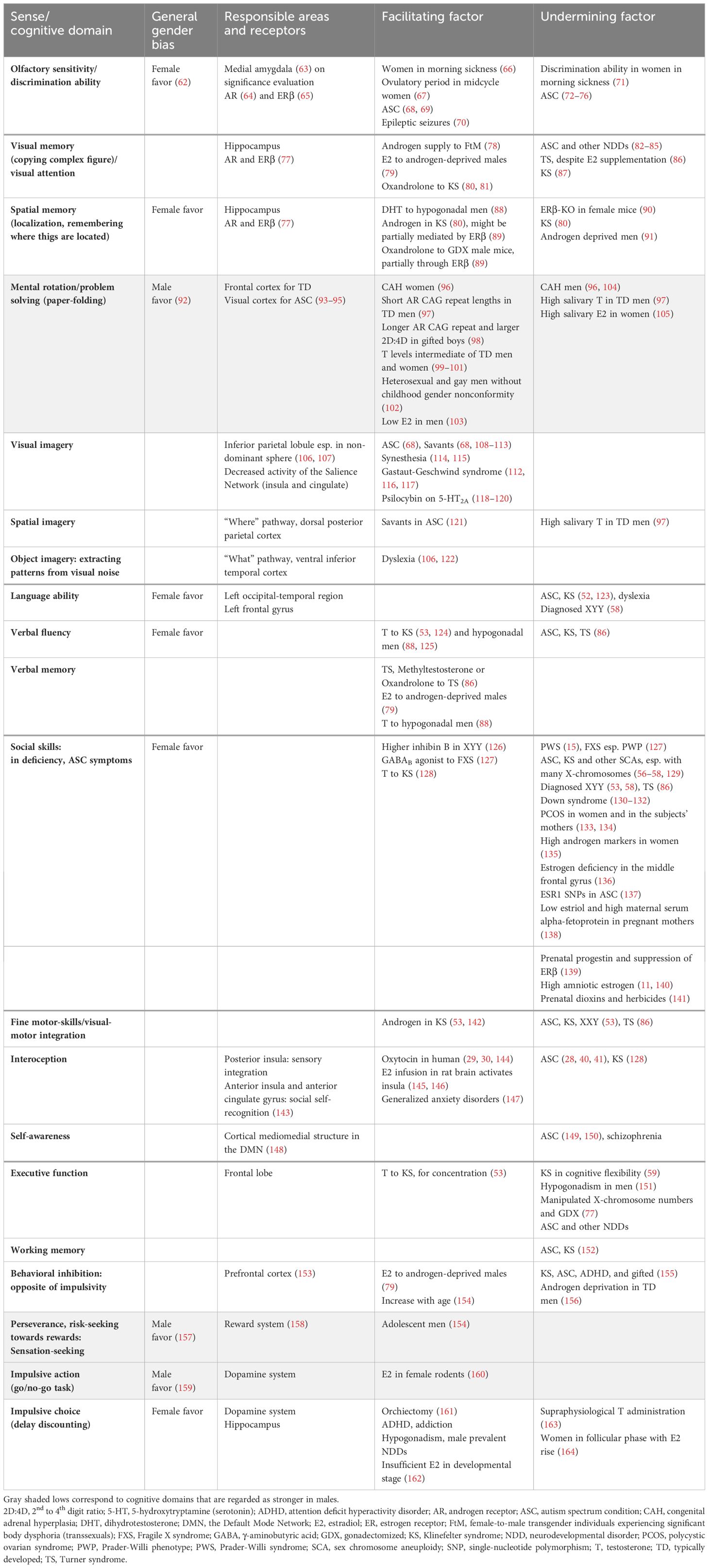
Table 1 Sex steroid effects on cognitive domains with observed sex-differences.
The XYY karyotype is rare, with approximately 1:1,000 males. They are characterized by tall stature, hypotonia, and cognitive problems; however, apparent symptoms are rare. Therefore, diagnosed individuals comprise a minority, with 0.7% (48), 5.9% (47), or at most 20% of XYY cases (168). Circulating testosterone levels are comparable to those in controls (48); however, infertility rate is higher than that in typical males (169, 170). There are sporadic reports of intersex states (testicular feminization) (171, 172) and transgender individuals (173) living as females. Among diagnosed individuals, cognitive and psychiatric profiles are similar to those of individuals with KS (53), but impairments in language and social responsiveness are more severe (58). To assess subtle differences in testicular function, particularly during prepuberty, inhibin B is a sensitive measure of testicular function, and lower inhibin B levels are associated with more autistic and problematic behaviors. Interestingly, pubertal rise in inhibin B is blunted, and prepubertal anti-Müllerian hormone levels are high, similar to those in KS. In XYY individuals, higher inhibin B or testosterone levels predict better cognitive, academic, and behavioral outcomes (126), contrary to the old expectation that XYY individuals are super-males and aggressive. Estradiol levels are low-normal (174, 175), and the osteoporosis rate is not high (48), suggesting that peripheral estrogen action is typical.
Individuals prenatally diagnosed with XYY show a higher-than-average IQ, in contrast to those who are postnatally diagnosed because of physiological/psychobehavioral problems. Postnatally diagnosed individuals with XYY show lower than average IQ and more ASC symptoms; however, physiological symptoms are comparable between the two groups. Although KS is characterized by shrunken testes, XYY males tend to exhibit macroorchidism, indicating a disrupted hypothalamic pituitary gonadal (HPG) axis (174).
Another genetic condition that connects hypogonadism and autistic cognitive predisposition is PWS. This is a genomic imprinting disease that lacks a paternally imprinted 15q11-q13 gene region due to mutations or maternal disomy. In contrast to the KS variations and XYY, but similar to XXXXY (57), the stature is short. Disruption of food intake regulation leads to obesity. Both males and females are since prenatally hypogonadal, adrenarche starts early, and the ASC prevalence rate is 11–80% (15). Personality problems are severe, with multiple learning difficulties and an IQ of 30–70 (176). Distinct from SCA is the inclination toward obsessive-compulsive disorders. Language delay is typical, as observed in the case of syndromic ASC, whereas special characteristics of PWS include the numbness to pain, vomiting stimuli, and temperature sensation. Additionally, individuals with PWS are very good at jigsaw puzzle (177). Therefore, locus 15q11-q13 has been considered to be responsible for savant syndrome (178), which includes relevant genes, such as gamma-aminobutyric acid type A receptor subunit gamma3 (GABRG3). Mutation of the gene affects the activity of the GABAA receptor subunit, hindering inhibitory signaling in response to GABA. However, this finding was not replicated (179), implying that this region only explains a certain subtype of savant syndrome. Interestingly, individuals with PWS tend to be fond of caring for animals and babies (180), suggesting that they are not indifferent to their interactions with animate agents.
Fragile X syndrome (FXS) is a triplet repeat disease caused by the extension of CGG repeats in the fragile X messenger ribonucleoprotein 1 (FMR1) gene, which resides in the 5′ end noncoding region on the X chromosome. This is the most common cause of mental retardation in males, and the most common single genetic cause of ASC. Affected individuals show an elongated face, large ears, other physical malformations, and difficulty in swallowing food. The FMR1 protein (FMRP) is one of the most abundant proteins in the brain and is crucial for synapse formation, regulating mRNA translation within dendrites. The mutation of FMR1 targets GABA receptor subunits and downregulates GABAA receptors. The protein is not synthesized when the number of CGG repeats exceeds 200, leading to obesity and behavioral problems. Anxiety, ADHD, and sensory hypersensitivity are frequent symptoms. The perseverance on topics is also frequent, sometimes meeting the criteria for obsessive-compulsive disorder. The abnormal enlargement of the testes begins at puberty. Significant to the current argument is the Prader-Willi phenotype (PWP) of FXS, which comprises 10% of all affected individuals; individuals with PWP have a syndrome similar to PWS, with heightened prevalence of ASC (54%) compared to 30% among males with FXS (127). The imprinting pattern of 15q11-q13 is normal; however, the cytoplasmic FMRP interacting protein 1 encoded by CYFIP1 in 15q11-q13 colocalizes and works together with FMRP in dendrites to form neuronal structures, and the downregulation of CYFIP1 results in this phenotype. In PWP, congenital hypogenitalism and hypogonadism emerge in 6 of 13 cases, with later development of macroorchidism in general (127). The full-scale IQ, including verbal and performance IQ ranges from to 36–49. In the molecular biological exploration of the common factors between FXS and ASC, the delay in synapse maturation, under-regulation of GABAA receptors and subsequent imbalance of glutamine/GABA neural systems are considered primary causes (127).
Additionally, chromosome 21 trisomy (Down syndrome) shows comorbidities with ASC (1–42%) (130–132) and ADHD (34%) (130). Individuals with Down syndrome show characteristic facial features, low IQs, hypotonia, and poor motor coordination. In males with Down syndrome, approximately one-fourth show cryptorchism and 10% show hypospadias (181). Infertility is prevalent, and hypogonadism is also reported to be high; however, sex steroid levels are relatively normal in the postpubescent population. Approximately one-third of the individuals show elevated LH or FSH levels, indicating primary gonadal dysfunction (181, 182). Some individuals show savant skills, such as music (183), but considered to be rare. Symptomology related to ASC has eluded research attention in this syndrome.
In the general population, registry-based studies in Sweden demonstrated that individuals of both sexes with hypogonadotropic hypogonadism or delayed puberty showed a significant increase in the prevalence of ASC, ADHD, and intellectual disabilities compared to matched controls (184). The impact of hypogonadism remained significant even after excluding individuals with chromosomal abnormalities from the analyses. Kallman syndrome is a genetic disorder that manifests with outcomes associated with congenital hypogonadism. Affected individuals show hypogonadotropic hypogonadism, anosmia, an elongated face, large ears, and an increased risk of osteoporosis, hearing loss, mental handicap, and schizophrenia (151). Gonadotropin-releasing hormone-1 (GnRH-1) neurons originate from the nasal placode, and atypical development at this earliest neural development stage leads to disorganization of HPG axis. Subsequently, several ASC risk genes require the facilitation by androgen or estrogen to fulfill their typical neurodevelopmental functions, as discussed in the next section. Additionally, testosterone (185) and estrogen (see Section 2.1.3) directly modulate GABAA receptor function, and their deficiency leads to destabilized excitation in the limbic system.
Androgen deficiency affects hypogonadal populations mainly on impairing communication abilities. Weaker activation of the dopaminergic reward circuit (see section 3.3) and the amygdala (see section 2.1.3) likely induces interpersonal anxiety and weaker interpersonal motivation. Impairment in executive control of behavior through weaker control of the prefrontal cortex (see section 3.3.2) and in spatial memory through hippocampal underdevelopment (3.2.1) are also possible outcomes. The effect of androgen is partly through AR and partly through ERβ in non-aromatized or after aromatized form.
2.1.2 Cognitive characteristics explaining ASC-savant traits and major possible target gene expressions triggered by sex-steroid deficiencyThe free energy principle is a broad theory in computational neuroscience that explains the formation of neural circuits and cognitive processes as the minimization of gaps between the expectation of signals and reception of input. As a comprehensive theory similar to Hebb’s rule, the free energy principle tries to explain how the information from stimuli is joined with the inner process representing phenomena in neural systems, and the closing gaps updates neural wiring. The principle was originally formulated to explain NDD phenomena, such as ASC and schizophrenia, and explains the characteristics of these two apparently different symptoms in the same model with different parameters. This principle has been demonstrated to be applicable to the decision-making process through the modulation of dopamine receptors (186), and to the learning process through cholinergic neuromodulation (187).
The neurodevelopmental hypothesis derived from the principle posits that a major causal factor of ASC symptoms is the disruption of GABA shift in early development (41) (Figure 2). The oxytocin-induced GABA shift changes GABAergic neuronal function from excitatory (depolarizing) to inhibitory (hyperpolarizing). Estradiol, especially its work on ERβ, is essential for organizing neural distribution in the developing brain (188, 189). Excessive estradiol delays GABA shift (190–192), and steroidogenesis dysfunction also delays this shift. For example, high concentrations of environmental disruptors, such as bisphenol A or bisphenol S hinder oxytocin function and GABA shift, likely by disrupting endogenous estradiol function (193). The delay or elimination of the shift leaves the brain in an immature state, leading to inflation of excitation/inhibition (E/I) ratio and a delay in closing the critical period postnatally, that is supposed to prune excess synapses, affecting neuronal plasticity (194). This leads to microstructural atypicality and hypoconnectivity of neural circuits around the amygdala, insula, and cingulate cortex, as well as psycho-behavioral atypicality. The GABA shift is an event that changes the brain from an organizational neural distribution phase to a synapse pruning phase in accordance with external sensory input (195).
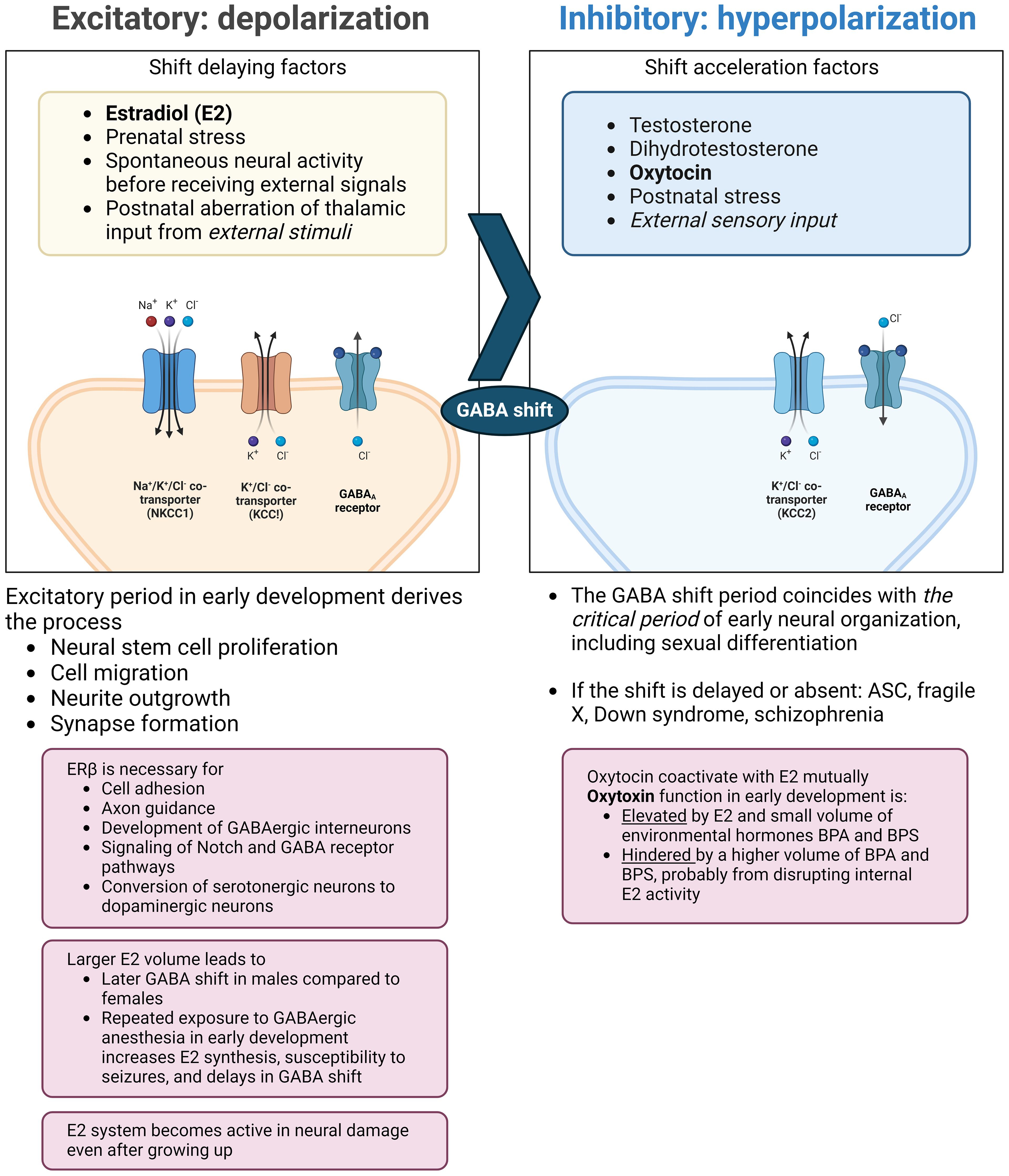
Figure 2 Endocrinological environment affecting the timing of GABA shift. Perturbation of ERβ activity hinders neural and glial cell development and adhesion. This also interrupts the function of oxytocin (right pane) and delays the timing of the GABA shift. The extended immature phase of the brain corresponds to weaker GABA inhibition and the skew to the serotonergic system compared to the dopaminergic system. Also, excessive estradiol doses by endogenous/exogenous causes a delay in GABA shift and can induce epilepsy, with possible subsequent distortion of steroidogenesis and functional pathways. ASC: autism spectrum condition; BPA: bisphenol A; BPS: bisphenol S; ER: estrogen receptor; GABA: γ-aminobutyric acid; KCC: K+-Cl- cotransporter; NKCC: sodium-potassium-chloride cotransporter. (Created with BioRender.com).
In contrast, the major psychological explanations of the cognitive characteristics of ASC, covering communication deficiency and higher performance in certain visual tasks, have been: 1) the Weak Central Coherence (196, 197); 2) the Enhanced Perceptual Functioning (EPF) (198, 199); 3) systemizing the brain through the EMB (7); and in a classic form, 4) the Geschwind–Galaburda hypothesis, which attributes the cause to the imbalance of hemispheric development (5, 6). The Geschwind–Galaburda hypothesis explains that higher prenatal androgen levels cause a delay in the development of left cerebral hemisphere, as manifested by an increase in non-right-handedness and atypical brain torque in individuals with ASC (200). The neurodevelopmental hypothesis indicates the order of development of these phenomena: the difficulty in closing gaps between internal and external perceptive stimuli and the expectation of received signals leads to non-optimal attenuation of perception, as manifested in hyper/hypoesthesia 2), and difficulty in summarizing perceived information 1), resulting in skewed strength/weakness in cognitive domains 3) and the skewed development of corresponding brain areas 4). Steroid deficiencies in early development and adulthood are possible causes of failure to attenuate neural signals. This could be manifested by overexcitation over primary perceptual input (201), overdevelopment of the visual cortex and primary sensory areas (38), glial cell atypicality perturbing synapses and neural circuit maturation (202–204). Delays in synapse maturation, reduced synapse pruning, and atypical neural networks of glutamine and GABA (205, 206) are hallmarks of ASC.
In higher-level cognitive processing, insular malformations are key to understanding the hindrance to integrating higher-order ego recognition and naming emotional experiences of self and others (36). The insula integrates multimodal sensations from the outside world and inner body into self-awareness. The posterior insula is an integral part of passing interoception to the brain, and it comprises visceral feelings and feedback from the autonomous nervous system. Disconnection from the interoception network leads to alexithymia (207). The inability to anticipate one’s own bodily responses leads to easy panicking and shyness (128). Although ERs are not densely distributed, estrogen infusion into the insula of the rat brain excites neurons by suppressing GABA release in this area and activates descending sympathoexcitatory pathways (145, 146). On the other hand, higher oxytocin concentrations correspond to stronger feelings of body ownership (29), sensitivity to the social environment (144), and emotional empathy in humans, possibly by strengthening the interoceptive network and attenuating physiological reactions to negative stimuli. Testosterone supplementation in individuals with KS improves communication and interpersonal skills, possibly by smoothing the expectation of perceptual input and helping integrate socially relevant information, thereby reducing anxiety about interacting with others (128, 208).
Major ASC-susceptible genes are modulated by estrogen and androgen, and steroid deficiencies can cause diverse cognitive symptoms. Forkhead box protein P2 gene (FOXP2), originally discovered in a family line with language developmental disorders, and its paralog, FOXP1, the fifth major risk factor for ASC and a common risk factor for other NDDs (209), are among them. FOXP2 is expressed in earliest-born cortical neurons in the subplate, and the protein binds to DNA to facilitate neural development and cell-type differentiation (210). Non-aromatizable androgens are necessary for normal expression and function of Foxp1 and Foxp2 in rats (211). Foxp2 is expressed in the sensory and motor-related cortices, cerebellum, and medial amygdala. Knockdown of Foxp2 in mice compromises social behavior processed in the medial amygdala, in dopamine-dependent manner (63). In female zebrafish, Foxp2 deficiency leads to disruption of the HPG axis (212), and FOXP2 is overexpressed in human prostate cancer cells (213). These results suggest that FOXP2 functions in the feedback on estrogen and androgen pathways.
Additionally, SH3 and multiple ankyrin repeat domains 3 (SHANK3) is another major gene factor against ASC, regulating neuronal and synaptic excitability (214). In line with SHANK1 and SHANK2, it functions on the N-methyl-D-aspartate (NMDA) and glutamate receptors, and is involved in dendritic spine maturation (215). SHANK3 is crucial for fixing and guiding the actin cytoskeleton of neurons before synaptic transmission; knockdown of SHANK3 reduces neuronal soma size, growth cone area, neurite length, and branch numbers (216). In macaque monkeys with the ASC-type mutant SHANK3, behavioral outcomes include sleep disturbances, motor deficits, repetitive behaviors, and social and learning impairments. Neuronal networks were also altered into hypo-connectivity in the Default Mode Network (DMN: see section 2.2.1) and local hyper-connectivity in areas including the somatosensory and posterior cingulate cortices (217). In human cell culture, dihydrotestosterone (DHT) increased the expression of SHANK by 35% and estradiol by 15%, indicating that both AR, ERα, and ERβ contribute to regulation (218). In contrast, Srancikova et al. found that testosterone downregulates SHANK1 and SHANK3, and that gene expression was lower in the hippocampus of male than of female rats (219).
In rodents, estrogen regulates sexual characteristics, puberty, and neurobiological reproductive systems through ERα. Contrary, estrogen modulates non-reproductive systems, such as anxiety, locomotion, fear, memory, and learning, through ERβ. If the action of estrogen on these neural systems during developmentally critical stages is insufficient, the typical development of neurons, synapses, and glial cells is hindered (220). The action of ERβ is not necessarily estrogen-dependent (188); the androgen metabolite androstanediol is also suggested to promote oxytocin function through ERβ (221). In human brain, areas with especially abundant ERβ are somatosensory cortex, hippocampus, thalamus, and cerebellum (222). Insufficient estrogen action is a possible causal factor for various psychiatric and NDDs that emerge with different sex ratios after puberty. In women, psychiatric syndromes, including schizophrenia, ASC, ADHD, and general anxiety disorders are exacerbated when estrogen levels are low (222).
2.1.3 Estrogen deficiency sometimes emerges with an increase in androgen markers in malesDifferentially expressed gene analysis across different phenotypes of ASC revealed that both androgen and estrogen signaling pathways are related to these conditions. Androgen signaling is associated with the emergence of a savant tendency (223, 224). Among ASC related genes, scavenger receptor class B type 1 (SCARB1) which codes a membrane protein regulating cholesterol usage in cells is also involved in sexual differentiation, and 5α-reductase type 1 (SRD5A1) gene controls cellular cholesterol intake and testosterone metabolism (224). However, these two genes have relatively small explanatory power among ASC-related genes (209). An apparent increase in steroid biomarkers may be secondary to a deficiency in sex steroid action during critical developmental periods and the subsequent disruption of steroidogenesis. Additionally, steroids functions not only trigger genetic expression cascades through classical actions on nuclear steroid receptors. Instead, many estrogenic neuromodulating mechanisms seem to depend on receptors on the membrane, which directly and rapidly modulate the strength of GABA or glutamate signal transmission in neurons (225, 226), and the activities of glial cells (98, 227–229), which guide neural extension, synapse modulation, and information transmission.
The G protein-coupled estrogen receptor (GPER) is a major estrogen membrane receptor expressed in primate LH-releasing hormone neurons in the olfactory placode and hypothalamus, modulating the HPG axis (230). Male HPG axis negative feedback loop is mainly controlled by estrogen instead of non-aromatized androgen in pituitary, suggesting the insufficient estrogen action behind the gonadotropin increase (231). GPER plays an important role in the control of spermatogenesis in the testes (231). Individuals with KS typically show hypergonadotropic hypogonadism, and their testicular tissue shows a 12-fold increase in GPER and a decrease in ERβ mRNA expression compared to control males, indicating insufficient estrogen control for testicular function in this population (232). GPER is expressed in both the central nervous system and peripheral tissues, including the cardiovascular system. Among children with ASC, serum GPER levels decreased with an increase in symptom severity. However, serum estradiol levels did not correlate with GPER levels (233).
During the developmental phases in men, excess androgen levels trigger negative feedback or are buffered within the high-normal range, which does not necessarily deliver unique developmental characteristics, as expected in women. For example, fetuses with congenital androgen hyperplasia (CAH) are exposed to androgens at supranormal concentrations in the adrenal glands because of a lack of metabolic pathways. This causes physiological and psychological masculinization (234, 235) only in females, but does not increase the odds of ASC, according to a recent meta-analysis (133).
When mothers suffer from polycystic ovary syndrome (PCOS) and androgen production is elevated, the conceived babies’ risk of developing ASC increases (134). Animal models suggest that in girls, the elevation of prenatal exogenous androgen levels is likely to induce postnatal upregulation of androgen production and activity, whereas in males, this induces a decrease in luteinizing hormone (LH) and underdevelopment of the testes (236). Estrogen concentrations in affected mothers and offspring are not consistently altered, but aromatase activity decreases in the placenta of mothers (237). In mice, the offspring of androgen-exposed mothers show downregulation of AR or ERα in a sex-dependent manner in the hypothalamus, hippocampus, and amygdala, showing anxiety symptoms. The expression of serotonergic and GABAergic genes tends to increase in a sex-dependent manner (238). A survey of gifted boys, who would have a common physiological background with savants reported that they had smaller 2D:4D finger ratios, suggesting the influence of higher androgens in the prenatal environment. However, salivary testosterone levels were significantly lower, and performance on reading in the eye test was poorer (239). This case might correspond with the case of offsprings who have been exposed to exogenous androgen in utero, while in male neonates, the association between amniotic testosterone levels and the 2D:4D ratio is questioned (240). Additionally, an initial report of smaller 2D:4D ratios in individuals with autism (241) was not replicated (12), or was limited to syndromic cases among men (242).
In an attempt to distinguish adults with Asperger’s syndrome from those typically developed with 24 serum biomarkers, only four markers were common between men and women. An increase in LH and free testosterone levels was observed only in women; among men, the main characteristics were higher levels of cytokines and other inflammatory measures (135). Geschwind et al. pointed that ASC and gifted are often suffering from autoimmune diseases (5). Autoimmune diseases are more prevalent in females compared to males in the general population. However, long-term aromatase deficiency induces autoimmune diseases in mice (243).
Congenital estrogen deficiency results from the deletion of the aromatase gene cytochrome P450 family 19 subfamily A member 1 (CYP19A1) or abnormal function of ERs (estrogen resistance). The PWP of FXS, who are at high risk of ASC (127), shows aromatase deficiency, with eunuchoid proportions, early onset metabolic syndrome, and oligozoospermia, similar to KS, in combination with cryptorchidism or macroorchidism (see section 2.1.1). Among various endocrinological diseases with the depression of estradiol levels, aromatase deficiency causes a large drop in estradiol levels; testosterone levels are low in some cases, and high in others (26). Mothers with fetuses affected by aromatase deficiency show virilization during the third trimester. Congenital estrogen deficiency in women leads to virilization of the genitalia, decreased estrogen levels, and increased androgen levels (27). In rats, prenatal exposure to synthetic progesterone in the form of oral contraceptives results in ERβ suppression in the amygdala and ASC-like behavior (244). Estrogen deficiency leads to insulin resistance, which hinders synaptic plasticity and dopaminergic function in the ventral striatum, thereby inducing anxiety and depression. Hypoactivation of the mesocorticolimbic and nigrostriatal dopamine pathways has been suggested to correspond to low social interaction motivation and stereotyped behavior (245).
Deficiencies in ERβ, CYP19A1, and ER coactivators in the middle frontal gyrus can be a direct cause of ASC (136). Acid-related orphan receptor alpha (RORA) is a transcription factor that induces aromatase expression through a feedback loop at sex steroid concentrations (246). In ASC population, RORA and aromatase expression are greatly decreased (247, 248). In a survey of the Japanese population, single nucleotide polymorphisms (SNPs) of ESR 1/2 were found to be related to the severity of symptoms in ASC. Human ER genes, ESR1 and ESR2, encode receptors that are homologues of ERα and ER β respectively, and ESR1 was concerned with the impairment of social interactions, and ESR2 with emotional regulation. However, ESR1/2 SNPs did not predict the severity of social communication problems, stereotypies, or sensory abnormalities (137). Among male and female transgender populations, which tend to co-emerge with ASC, many gene variants correspond to estrogen signaling pathways in sexually dimorphic brain areas, but none to androgen pathways (249).
In the prenatal environment, low concentrations of unconjugated estriol (uE3) and high concentrations of maternal serum alpha-fetoprotein (MSAFP) in the maternal serum increased the odds ratio of ASC prevalence in offspring (138). A low uE3 concentration is an indication of insufficient production of adrenal steroid (dehydroepiandrosterone and others) in infants, and MSAFP suppresses estrogen activity. Additionally, prenatal exposure to progestin, which is prescribed to prevent threatened miscarriage, has been reported to suppress the expression of ERβ in the fetal brain, thereby increasing the risk of ASC in rat experiments and epidemiological studies (139).
The above evidence suggests that one of the main factors that induce ASC is the depression of estrogen action; if the potential for steroidogenesis in the fetus is intact, the androgen production rate would be increased to compensate for estrogen deficiency. In another case, an increase in exogenous androgen circulation downregulates endogenous androgen genesis in the fetus, that will cause estrogen deficiency subsequently; however, using a Danish cohort sample, Baron-Cohen et al. showed an increase in various estrogen and progesterone concentrations in amniocentesis fluids of boys with ASC (11, 140).
In addition to syndromic ASCs that lack steroid hormones during early development, there are known cases of externally caused shortages of sex steroids that induce ASC. The responsible environmental disruptors are dioxins and herbicides (141). An analysis of umbilical codes suggests that exposure to high levels of dioxins suppresses androgen production in male fetuses (250). Bisphenol A (BPA) is a blocker of AR (251) and ER, affecting the expression of multiple ASC related genes. Bisphenol A exposure in pregnant rats increased neurite length and the number of neurite branches in offspring of both sexes, while an increase in neuronal cell death, the impairment of neuronal development in the hippocampus and learning ability were observed only in male offspring (252). The prefrontal cortex of adult Long-Evans rats prenatally exposed to high concentrations of BPA showed an increase in the number of neurons and glia in layers 5/6, but only in males (253). In contrast, an epidemiological test using a public human cohort demonstrated that the effect of BPA on ASC susceptibility was more apparent in girls (254).
Studies have reported the disruption of steroidogenesis in individuals with ASC, particularly alterations in metabolic pathways before steroids are converted into sex hormones (255). Along with estrogen, neurosteroids such as the progesterone metabolite allopregnanolone, are crucial for neural cell proliferation, migration, myelination, synapse formation, and modulation of GABAA receptors in the cerebral cortex, thalamus, and hippocampus (256, 257). This function persists from the earliest stages of brain development to maturity. Androgens show similar functions or work in consort with estrogen, but their importance in early neural development and cognitive modulation seems to be secondary, as direct pharmacological evidence of the effects of developmental androgen alteration is limited. Such reasoning resolves the discrepancy, especially in affected males, where researchers have largely failed to find an increase in androgen markers prenatally or postnatally (133, 255).
One epidemiological support for the early idea that an abnormal increase in steroidogenesis causes ASC comes from the fact that peripubertal (15–19 years) individuals with ASC have a higher incidence of genital/ovarian cancer. Mothers of individuals with ASC also frequently experience sex hormone-responsive cancers (258). The increased vulnerability to disease, likely derived from the abundance of sex steroids, is more evident in female ASC than that in male ASC cases. Some ASC-related genes, such as FOXP1 and copy number variation, are known to suppress tumors or cancer if they are intact, requiring further examination to explore how such a genetic background interacts with steroid excess contributing to cancer risk.
2.2 Extreme success in “masculine” artistic-academic fields inversely correlate with masculinity indices among men2.2.1 Connection between developmental disorders, savant/gifted, and atypical sexual differentiationApproximately 50% of individuals with savant ability are from the ASC population (259), and the emerging ratio of sex differences is approximately three men to one woman (260). Savant ability is independent of total IQ scores. In the general population, prodigious savants who show distinguished talents that are difficult to interpret with common sense, are very rare, with only approximately 50 individuals worldwide. If the definition is expanded to talented savants who show some distinguished abilities relative to other domains of talent of their own, the prevalence among the ASC population is estimated to be from 28.5% (260) to 42% (261) or nearly 50% (262). In the early stages of academic notice, savant art was considered as an emotionless repetition of the gross amount of memory without original creativity. This notion was later corrected; savant art has its own originality, and is considered a model of the root of creativity (262, 263).
In contrast, gifted individuals are defined as “those who show exquisite talents in one or several fields (logical thinking, learning ability, etc.), or those who show abilities of top 10 scores of a population (264).” The greater the extraordinary ability, the greater tends to be the developmental unevenness across cognitive domains and social maladaptation. The behavioral output of gifted individuals is often difficult to distinguish from or co-exists with ADHD, obsessive-compulsive disorder, ASC, schizophrenia, and avoidant personality disorders (155). Whether an individual is judged as savant or gifted is largely influenced by the academic background of the report.
Using the Bayesian explanation of the free energy principle, hypopriors (having fewer internal models prior to processing stimuli) can explain hypersensitivity to sensory input, immunity to being distracted by contextual information (better at copying impossible figures), and incompetency in communication dependent on summarized inference (265) (Figure 1). Therefore, the EPF model in combination with the free energy principle does not require supposing a lack of empathic motivation in the first place, but suggests that the characteristics of perceptual processing are the basis of both unevenness in the strength of cognitive domains and communication inaptness.
The main premise of the EBM theory is that higher androgen action on the prenatal brain increases ASC traits between and within sexes. However, ASC traits are more observable among hypoandrogenism conditions, contrary to expectations, except for females with PCOS and offspring of mothers with PCOS (133). The effects of maternal PCOS on offspring are under debate, with complications from epigenetic effects, genetic inheritance, insulin resistance (266, 267), and the effects of metformin administration on mothers (268). Additionally, Baron-Cohen et al. admit that MRI voxel-based morphological ASC-TD brain differences within sex are not parallel between sexes, and the difference within men is especially discordant with TD sex differences (13). They also reported in adults, blood serum biomarkers, such as high LH and free androgen index could distinguish between women with Asperger’s syndrome and controls; however, the same marker set did not have discrimination power for men (135).
The DMN is the functional connection of brain areas involved in self-referential mind wandering, which becomes active when a person is not focused on performing tasks. Researchers have become increasingly interested in the DMN because of its ability to discriminate atypical cognitive statuses. Baron-Cohen et al. compared the connectivity strength of the DMN between sexes in TD and within sexes across ASC and TD groups. They reported that TD males and individuals with ASD had weaker connectivity in the DMN (269). In contrast, in an analysis of whole-brain connectivity without presupposed theories, the connectivity between social brain areas (fusiform gyrus, superior temporal sulcus, inferior parietal cortex, insula, and posterior cingulate cortex) and other areas was stronger in female ASC (male-like), but weaker in male ASC (female-like) (270). They argued that this result supports the atypical sexual differentiation theory of ASC genesis instead of the EMB theory.
Do gifted individuals tend to be hypermasculine or gender neutral? Several studies support the latter tendency. The fields of interest and strengths of gifted individuals tended to be gender-neutral instead of following stereotyped gender categories (155, 271). Their gender identities also tend to be neutral (272). Another survey showed that neutral gender identity was more evident among gifted women than in men (273). Additionally, more students in gifted classes recognized themselves as sexual minorities than their non-gifted counterparts (274).
3 Expectation mismatch on perceptual input will lead to uneven development of perceptual processing and difficulty in information integration3.1 Oversensitive perceptual input as the basis of weak central coherence, extreme systemizing, and communication deficiency3.1.1 Hyperacuity and overexcitability over sensory input likely start at early developmentWidely observed cognitive underpinnings among ASC and other NDDs are the dominance of primary and local sensory information processing, the weakness of abstracting from that information, and having a global view. They also have difficulties combining information of different modalities over time. The peculiarities of vision perception have been extensively studied. This can lead to excellence in information processing in certain domains, such as in savants, and at the same time, difficulties in real-time responses needed for communication (201, 275). Perceptual distinctiveness observable daily is the inflation or suppression of senses in response to external stimuli (149, 276), a characteristic shared by the gifted (277). The high excitability of neurons because of insufficient suppression by GABA, and increased glutamine concentrations are considered physiological factors (201).
Brain development starts from lower-level sensory areas; subsequently, integration functions in the association areas and prefrontal lobes develop over a long period. The peculiarities of developmental trajectory in individuals with ASC are observable immediately after birth; their brain size is larger than that of TD individuals in the early stages of life (278), especially in the visual cortex and primary sensory areas, followed by the inferior temporal cortex (38) (Figure 1). Dominance of local visual processing seems to start in infancy (279), indicating that peculiarity in perception begins at the earliest development of primary sensory receptive fields, inducing the overdevelopment of certain perception-oriented information processing neural network traits, and neural excitability (280). Hypersensitivity of receptive fields and sensory processing in ASC individuals seems to be underlined by the peculiar density in neural cell packing; in ASC postmortem brains from infancy to young adults, mini-columns in the cerebral cortex were narrower in Brodmann area 3 (primary sensory area), 4 (primary motor area), 9 (prefrontal association area), 17 (V1), 21 (temporal visual association area), and 22 (temporal auditory area) (202, 281). In other specimens, ASC did not differ in cell structure in area V1 (Brodmann area 17), and narrowing of the mini-columns was most prominent in the ventral and orbitofrontal prefrontal lobes (282).
The function of estradiol on ERβ seems to be critical for early neural/glial distribution and closing the immature phase shifting to stimuli-dependent synapse modification (Figure 2). The time phases and pathways of neural maturation differ across perception modalities. In embryonic stem cell lines in which SHANK3 was mutated into the ASC type, olfactory placodal neurons first emerged at the earliest stage of neurogenesis, and then developed smaller cell bodies and more and longer neurites compared to those in controls. These shifts were not observed in cortical neurons, except for the shortening of neurites (283). Similar changes were observed in the auditory areas, but in the opposite direction, with a wider interval between mini-columns compared to that in controls in the primary auditory and association fields. This difference was particularly significant in the primary auditory field in younger populations (284).
As stated previously, hyperacuity in the visual senses appears to be underlined by differences in neural cell structures. Notable characteristics of individuals with ASC include larger numbers of near-distance neural connections (285), a larger proportion of intra-hemispheric in contrast to inter-hemispheric neural connections, an inclination to excitation in the neural E/I balance, and heterogeneity of development between brain areas. The incoherence of processing and communication speed across modalities makes it difficult to combine information in real-time, resulting in the creation of unique information-processing networks that detour neurotypical processing in language areas, which may manifest as a unique brain torque (200) or non-right-handedness (286) in parts of the population.
3.1.2 Visual imagery and synesthesia as the bases of savant abilityTypically appearing cognitive skills in savants are categorized as follows: 1) calendar calculation; 2) music; 3) art; 4) mathematical and number skills; 5) mechanical or spatial skills; and 6) “other obscure skills” which include exceptional multilingualism, sensory discrimination abilities, synesthesia, and knowledge in specific fields (121, 287). For ASC individuals, Temple Grandin grouped characteristic cognitive styles into the following: 1) visual thinkers; 2) music and math thinkers (pattern thinkers); and 3) verbal logic thinkers (288). Distinct cognitive strategies observable in high-functional ASC, savants, and gifted are calculation, architecture, and art creation by manipulating vivid visual imagery (68, 108–113). Enhanced pattern detection is likely to develop through exceptional sensory acuity and veridical mapping across isomorphic structures (289).
Individuals with ASC tend to preferentially use the visual cortex, in contrast to neurotypicals who use the prefrontal cortex to solve visuospatial tasks, such as mental rotation, under experimental conditions (93, 94). Furthermore, even in non-spatial tasks, such as the N-back working memory task which is typically processed verbally in neurotypicals, ASCs tend to use the right hemisphere and posterior cerebral regions, including the inferior temporal area and occipital lobe, indicating that they solve tasks visually (95). Inferior temporal gyrus comprises “what” ventral pathway in visual processing and responsible for object imagery handling color, texture, and patterns. The area stores single cells which index long-term memory, connecting semantic significance to the images, and also processes letters and manipulates numbers. Among individuals with ASC, visual imagery itself, focus on detail, and low communication ability were not related to savant abilities; however, high spatial mental imagery predicted a larger number of savant abilities (121). Spatial imagery is processed in the dorsal visual pathway leading to the occipito-parietal regions.
Synesthesia is another key to understanding savant phenomena through atypical sensory processing. In synesthesia, the senses of the different modalities are jointly perceived. Synesthesia is relatively common in ASC populations and closely associated with the emergence of savantism (290). Mental imagery ability is closely associated with sequence-space synesthesia, leading to savant abilities, such as calendar calculation (114, 115). The inferior temporal cortex and areas adjacent to the fusiform gyrus are responsible for grapheme-color synesthesia (291). In contrast, a survey of a larger population reported that the emergence of synesthesia was independent of the strength of visual imagery, and that individuals with weak visual imagery could possess synesthesia (292). Connectivity in the superior parietal or frontal cortex is responsible for the synesthesia irrespective of its subtype (291). Absolute pitch recognition is an aspect of veridical mapping and sound-related synesthesia that sometimes hinders language recognition, which requires the abstraction of information from similar phoneme patterns (289, 293).
In summary, the endophenotype connecting the cognitive characteristics commonly observed among ASC and other NDDs, savant and gifted individuals is heightened sensory sensitivity (121). The overdeveloped cortical parts in ASC high-risk infants overlap to those in male-to-female transgender individuals experiencing significa
留言 (0)