
記住我










The importance of studying sex differences is today well recognized in many research fields, including neuroscience (Jazin and Cahill, 2010; Zagni et al., 2016; Golden and Voskuhl, 2017; Choleris et al., 2018). However, no studies have yet specifically addressed sex differences in multiple human embryonic stem cell lines (hESC). Models of stem cell differentiation are invaluable for this kind of research because they allow the study of genetic sex differences in the absence of sex hormones and their effects. Sex differences can have critical implications for the tissues they affect. In the developing nervous system, they have the potential to influence, e.g., the number of neurons, glial cells, synapses, and their organization. As a consequence, neural networks may develop that respond to stimuli in sex-specific ways (Arnold and McCarthy, 2016). Sex differences appear to be particularly relevant for several neurological diseases. Autism spectrum disorder, Tourette syndrome, and attention-deficit/hyperactivity disorder, for example, exhibit sex biases in symptoms, onset, and prevalence (Boyle et al., 2011; Loke et al., 2015; Tesic et al., 2019). These disorders are believed to have a neurodevelopmental origin, and it is likely that sex differences in early brain development act as a major contributor to the sex-biased development and manifestation of these diseases. Currently, sex differences in the human brain are largely attributed to the effects of sex hormones during critical periods of fetal development and adolescence (McCarthy, 2009; Arnold and McCarthy, 2016). However, an increasing body of evidence demonstrates that not only hormones alone play a role but also genetic factors, such as sex chromosome dosage compensation (Raznahan et al., 2018; Sidorenko et al., 2019; Warling et al., 2021), and differences in gene expression contribute to sex differences in the developing human brain (Carruth et al., 2002; Corre et al., 2016; Arnold, 2017; Gegenhuber and Tollkuhn, 2020; Cabrera Zapata et al., 2022). Despite the growing attention that research in sex differences receives, only a few studies specifically aim to investigate sex differences in embryonic stem cells, and to our knowledge, none of them include more than 1 cell line of each sex. Particularly, the role of sex-biased gene expression in early neuron development remains unclear. To address this gap, we collected four male and four female human embryonic stem cell lines, differentiated them into a mixed neuronal population over 37 days, and analyzed their gene expression profiles for sex differences using both total RNA sequencing and qPCR analysis. Our results reveal the presence of gene expression sex differences in all investigated stages of neuronal differentiation, but most pronounced after 37 days. We identified a number of candidate genes with potential contributions to sex-biased neuronal development. Finally, we demonstrate how overexpression of X-linked gametologous genes that escape X chromosome inactivation (XCI) is compensated by the expression of corresponding Y-linked homologs.
In summary, our research addresses a critical gap in understanding how sex differences in gene expression may influence early neuronal development. These findings could have implications for understanding the basis of sex-specific responses in the nervous system and their relation to neurological disorders.
MethodsCell linesThe human embryonic stem cell lines WA14 p33 (XY) (Thomson et al., 1998) and WA09 LT2e-H9CAGGFP p80 (XX) (Macarthur et al., 2012) were acquired from WiCell, Madison, Wisconsin. All other lines, HS975 (XX) p16, HS980 (XX) p14, KARO1 (XX) p12, HS1001 (XY) p22, HS983a (XY) p12, HS401 (XY) p33 were acquired from the Karolinska Institute Human Embryonic Stem Cell Bank (Main et al., 2020). The cell lines were tested for common karyotypical abnormalities with the STEMCELL hPSC Genetic Analysis Kit (#07550). None of the lines showed signs of abnormalities (Supplementary Figure S8).
Cell cultivation and differentiationThe hESC lines were cultivated as monolayers in mTeSR1 plus medium (STEMCELL), on 6-well cell culture dishes (Sarstedt 83.392) coated with Matrigel (Corning). The cultures were maintained in 5% CO2 and a temperature of 37°C. Dissociation of the cells was performed using a Versene (ThermoFischer 15040066) incubation of 8 min at room temperature. Each differentiation experiment was conducted in triplicates for each cell line and the protocol was identical for each cell line.
The neural differentiation protocol was based on (Chambers et al., 2009; van de Leemput et al., 2014) and was aimed to create a population of mixed neurons. The initial neural induction was performed on poly-L-ornithine (20 μg/mL, Merck P3655-50 MG) and laminin (20 μg/mL). Each we of a 6-well plate was coated with 1 mL poly-L-ornithine, incubated over night at 8°C followed by an incubation for 1 h at 37°C. Afterwards, the wells were rinsed with cell culture grade H2O and coated with 1 mL laminin solution, followed by the same incubation conditions as described before.
Differentiation was induced by dual SMAD inhibition using KO-DMEM/F12 and 1X knockout stem replace (KSR, Thermo Fisher 10828028) together with 10 µM SB-431542 (TargetMol T1726) and 0.1 µM LDN-193189 (TargetMol T1935) for 9 days. On day 3, an additional 3 µM CHIR-99021 (TargetMol T2310) and 1 µM cyclopamine (TargetMol T2825) was added and kept until day 17 of differentiation. On day 5, a transition from KSR medium to a N2 supplement (ThermoFischer 17502048) based medium in DMEM/F12 was performed in 25% steps per day. On day 6 the cells were split 1:3 and reseeded on Matrigel coated plates. On day 18, medium was changed to neurobasal medium with 1X B27 (ThermoFischer 17504044), 55 µM β-mercaptoethanol (ThermoFischer 31350010 µM), 10 µM cAMP (ApexBio B9001), 200 µM ascorbic acid (Merck A4403), 20 ng/mL GDNF (Peprotech 450–150), 20 ng/mL BDNF (Peprotech 450–02) and 50 ng/mL β-NGF (Peprotech 450–01). Each medium also received 2 mM glutamax (ThermoFischer 35050061) and 0.1 mM non-essential amminoacids (ThermoFischer 11140035). Medium was replenished every day until day 17 and afterwards every second day. For the start of the differentiation 150.000 cells were seeded into the coated dishes together with mTeSR1 plus medium. After 24 h of incubation, medium was changed to KSR medium to start the differentiation.
The cell media in this experiment, such as mTeSR+ and NB, as well as their supplements KSR, N2 and B27 can contain hormones such as insulin, progesterone and triiodothyronine. These hormones have proven to be essential for the maintenance and differentiation of stem cells. Except from the presence of progesterone, we have no evidence for the presence of other steroid hormones in the cell cultures.
RNA sampling and sequencingRNA sampling for qPCR as well as for RNA sequencing was performed in the following way: cells were washed with 2 mL PBS, thereafter 1 mL TriZol (ThermoFischer 15596026) was used for lysation. The lysate was passed six times through a syringe (BD Microlance 3 (27G ¾ (Nr.20) 0.4 × 19 mm) Ref: 302200) to promote cell lysis. The TriZol phases were separated using chloroform and the RNA phase was purified with the PureLink RNA Mini Kit (ThermoFischer 12183018A). Subsequently, the RNA was depleted of DNA using the DNA-free DNA Removal Kit (ThermoFischer AM 1906). All steps were executed according to manufacturer’s instructions.
Cell lysates were obtained from one 6-well-plate well before differentiation (D0) and after start of differentiation (D4, D9, D17, D27, D37) using 1 mL TriZol (ThermoFischer 15596026). TriZol phases were separated using chloroform and the RNA phase was purified with the PureLink RNA Mini Kit (ThermoFischer 12183018A) according to manufacturer’s instructions. Subsequently, the RNA was depleted of DNA using the DNA-free DNA Removal Kit (ThermoFischer AM 1906).
RNA samples were sent in triplicates for sequencing to the SNP&SEQ Technology Platform in Uppsala where the RNA quality was verified using a Bioanalyzer (RIN ≥8). Samples were then treated with the TruSeq Stranded Total RNA and Ribo-zero Gold kit (Illumina). The treatment preserves strandedness of transcripts but depletes the samples of cytoplasmatic and mitochondrial ribosomal RNA. Sequencing was performed on the NovaSeq instrument with an S4 lane (Illumina) resulting in approximately 27.7 million 150 bp pair-read read-pairs per sample. The data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus (Edgar et al., 2002) and are accessible through GEO Series accession number GSE249035 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE249035).
Differential gene expression and gene set enrichment analysisThe samples were aligned to GRCh38_r96 (Schneider et al., 2017) with Hisat2 (Kim et al., 2019) and sequencing reads were assigned to genomic features using featureCounts (Liao et al., 2014). Quality control was performed with FastQC and MultiQC (Ewels et al., 2016). The differential gene expression analysis was performed with DEseq2 (Love et al., 2014). The two factors “Sex” and “Time” were joined and the contrast function was used to compare the differential gene expression between and within the biological sexes (XX to XY) at each timepoint (D0, D4, D9, D37), Figure 3 A-D. Differentially expressed genes were then filtered for a minimal average of 50 counts in any of the cell lines of one sex, and for a fold-change bigger than 0.5 with an adjusted p-value lower than 0.05. The count limit of 50 accounts to approximately 2 CPM in our data and is considered stringent. The value was selected after inspection of the histogram of average logCPM values and an appropriate threshold was selected between the low-CPM peak representing non-expressed genes and the high-CPM peak representing expressed genes.
To acquire genes that are truly sex different, a stringent filter was implemented. It consisted of the standard parameters: fold-change (≥0.5) and adjusted p-value (≤0.05), but also includes the coefficient of variation for genes within cell lines of each sex (≤0.5) and a minimal difference between the expression of cell lines of the opposite sex (≥20% difference in gene expression). The filter kept only those genes that are expressed at a similar level within cell lines of the same biological sex and if none of the cell lines of one sex expresses the gene of question as high as any cell line of the other sex. For this, the coefficient of variation for genes within the cell lines of each sex was required to be lower than 0.5, to remove genes that are highly variable in expression and hard to predict. Additionally, the difference in gene expression between the cell lines of the opposite sex had to be higher than 20%. The latter required all 9 comparisons (3 male vs. 3 female lines) to display a difference of at least 20% in DEseq2 normalized counts. To investigate genes that contribute significantly to the neuronal differentiation, XX and XY counts were combined and the timepoints were contrasted (D0 to D4, D0 to D9 and D0 to D37), Figures 3E, F.
For the gene set enrichment analysis, the GSEA/MSigDB software was used, software version 4.2.3 and molecular signatures database version 7.5.1 (Mootha et al., 2003; Subramanian et al., 2005). Preranked GSEAs were performed using the stat parameter of the DEseq2 analysis, which represent the difference between the selected comparison including the magnitude of fold-change and p-adjusted. The C5 ontology gene sets were used consisting of gene sets for biological processes (BP), cellular components (CC) and molecular functions (MF). The resulting data was used to create enrichment maps using the Cytoscape software version 3.91 (Shannon et al., 2003) and the auto annotate app for cluster annotation based on gene set description. Enrichment maps were created using an FDR q-value cut-off of below 0.1 and a perfused force directed layout based on the similarity coefficient of gene sets.
qPCRFor the gene expression analysis by qPCR, cDNA was synthesized from the purified and DNA free RNA using Maxima H Minus Reverse Transcriptase (ThermoFischer EP0752) with anchored Oligo (dT)20 Primer (ThermoFischer 12577011). The following protocol was used for cDNA synthesis in a 15 µL reaction: 500 ng rDNAse-treated RNA, 1.25 μg/μL Oligo (dT)20 Primer, 1 µL dNTP mix (10 mM each, Thermo Fisher R0191), 4 µL RT buffer, 0.5 µL RiboLock RNAse inhibitor (ThermoFisher EO0381), 0.5 µL Maxima RT enzyme and RNAse free water was used to top up to the desired volume. Incubate at 50°C for 30 min, terminate at 85°C for 5 min. A NanoDrop spectrophotometer was used to assess the RNA amount and quality. RNA integrity was measured using gel electrophoresis and comparison of 28S/18S ribosomal RNA ratios. The qPCR analysis was performed on the Applied Biosystems 7500 Real-Time PCR System using PowerUp SYBR Master Mix (Applied Biosystem A25742) and standard cycling mode. For every reaction 1 ng of cDNA was used with 7.5 μL of SYBR master mix and 1 μL of 10 mM forward and reverse primers. Molecular grade H2O was added for a total volume of 15 μL. qPCR measurements were taken in triplicates. The thermal cycling profile was selected according to master mix recommendations (1 cycle UDG activation, 50°C, 2 min; 1 cycle activation of Dual-Lock DNA polymerase, 95°C, 2 min; 40 cycles of denaturation, 95°C, 15 s, and annealing/extension, 62°C, 1 min), all runs were performed at an annealing temperature of 62°C. Amplification cycles were repeated 40 times followed by a melting stage, serving as a diagnostic tool to assess amplicon length. Primer sequences can be found in Supplementary Table S9.
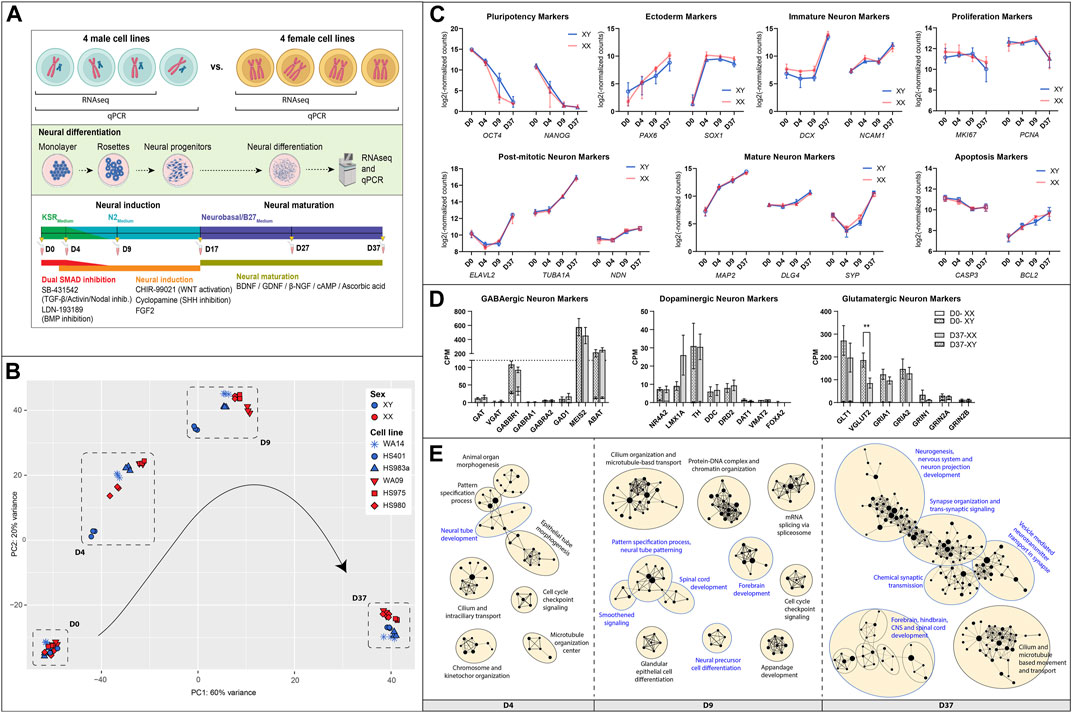
ResultsSimilar neuronal differentiation of male and female human embryonic stem cellsTo identify potential sex differences in the development of human embryonic stem cells (hESC), we differentiated four male and four female-derived hESC lines towards a population of mixed neurons. The differentiation protocol utilizes adherent cell culture combined with small molecules for neural induction and maturation, as described in detail in the Methods section. The cells were differentiated for a total of 37 days, and samples were collected at six timepoints: D0, D4, D9, D17, D27, and D37 (Figure 1A). Six cell lines were analyzed by total RNA sequencing at four timepoints (D0, D4, D9, D37), and all 8 cell lines were analyzed via qPCR at all six timepoints (Table 1).
Figure 1. Similarities between male and female hESC in neuronal differentiation: (A) Overview of the experimental procedure: Four hESC lines of each sex were differentiated for 37 days to a population of mixed neurons induced by small molecules in 2D culture. Bulk RNA sequencing and qPCR. (B) A PCA analysis of expressed genes displays similar gene expression patterns at D0, some inter-cell line differences during early differentiation, and a sex-biased separation at D37 (XY: WA14, HS401, HS983a, XX: WA09, HS975, HS980). (C) Marker gene expression for pluripotency (OCT4, NANOG), ectoderm (PAX6, SOX1), immature neurons (DCX, NCAM), mature neurons (MAP2, DLG4, SYP), post-mitotic neurons (ELAVL2, TUBA1A, NDN), as well as for proliferation (MKI67, PCNA), and apoptosis (CASP3, BCL2) show a similar expression in male and female cell lines. (D) Markers for specialized neurons (GABAergic, Dopaminergic, Glutamatergic) display a premature state of the neurons after 37 days of differentiation. (E) A gene set enrichment analysis at D4, D9, and D37 reveals an upregulation of biological processes implicated in neuronal differentiation in all cell lines. The GSEA was summarized in an enrichment map. Each dot represents a gene set; the size of the dot corresponds to the size of the gene set. If the dots are connected or in close proximity, the more the gene sets are overlapping, and the closer related are their associated biological functions. Gene sets were grouped by a common GO-term classification and named after an overarching biological function. Biological functions implicated in neuronal development are highlighted in blue. Gene sets are filtered by FDR q-values <0.1. Figure 1A created with BioRender.com.
Table 1. hESC lines and gene expression analysis method and timepoints.
A principal component analysis of expressed genes revealed that male and female cell lines show similar gene expression at D0 but display cell line-specific differences at D4 and D9 (Figure 1B). After 37 days of differentiation, the differences decreased, and male as well as female cell lines show similar gene expression. However, we noticed a clear sex-specific separation between them according to principal component 2 (PC2).
In general, the cell lines of both sexes have differentiated similarly towards neurons, displayed by very similar expression dynamics of marker genes for pluripotency (OCT4, NANOG), ectoderm (PAX6, SOX1), immature neurons (DCX, NCAM1), mature neurons (MAP2, DLG4, SYP), post-mitotic neurons (ELAVL2, TUBA1A, NDN), as well as for proliferation (MKI67, PCNA) and apoptosis (CASP3, BCL2), Figure 1C. In addition, the pluripotency state of all cell lines was investigated and identified as primed by the overexpression of naive over primed pluripotency marker genes at T0, Supplementary Figure S1. To estimate the replicability of the differentiation protocol, the experiment was repeated two more times, and the expression of NES, PAX6, and DCX was measured using qPCR. We observed consistent gene expression levels in each repetition, indicating low variability and a robust differentiation protocol. Additionally, we identified sex differences which were not detected in the RNAseq data for NES at D9 (1.8 FC, p < 0.0001), in PAX6 at D37 (1.3 FC, p < 0.05), and in DCX at D17 (2.2 FC, p < 0.0001), Supplementary Figure S2.
An investigation of marker genes for specialized neurons has revealed that male as well as female cells similarly differentiated to premature neuron-like cells after 37 days (Figure 1D). For example, the premature state of GABAergic neurons is suggested by the high expression of GABBR1 (GABA type B receptor unit) combined with high expression levels of MEIS2 and ABAT (both GABAergic precursor markers), while other GABAergic markers, such as the type A receptor GABRA1/2 or the neurotransmitter transporters SLC6A1 (GAT) and SLC32A1 (VGAT), as well as the rate-limiting enzyme for GABA synthesis GAD1 are expressed very low or not at all. Similarly, dopaminergic markers NR4A2 and LMX1A, which are transcriptional regulators for the development of DA neurons, indicate the presence of neurons that are not fully mature. However, the expression of the catecholamine precursor L-DOPA-synthesizing-enzyme tyrosine hydroxylase (TH) and the L-DOPA to dopamine converting enzyme dopa decarboxylase (DDC), as well as the dopamine receptor D2 (DRD2), point to an, at least, early post-mitotic state of some dopaminergic neurons. The dopamine transporters SLC6A3 (DAT1) and SLC18A2 (VMAT2), as well as the transcription factor FOXA2 that supports maintenance of mature DA neurons, are expressed at low levels or not at all. As for the glutamatergic neurons, we can see high expression of the glutamate transporters GLT1 and VGLUT2, as well as for the glutamate AMPA type receptors subunits GRIA1 and GRIA2. Glutamate NMDA type receptors were also expressed, albeit at a moderate to low level (data not shown). Interestingly, the VGLUT2 gene shows a sex difference and is higher expressed in male cells than in female cells after 37 days of differentiation (p = 0.009 **).
Gene set enrichment analyses (GSEA) also revealed a similar differentiation of male and female cell lines. We performed GSEAs for each sex separately (e.g., XY D0 to D4 and XX D0 to D4) and compared them. We found that the upregulated gene sets representing biological processes in the cells during the differentiation experiment are almost identical with only minor variations between the sexes (data not shown). A simple way to visualize this is by performing a GSEA with pooled data from all cell lines. This has resulted in a single enrichment map in which gene sets will only appear if they are upregulated in all cell lines, male and female (Figure 1E). Each dot in the map represents an upregulated gene set. Upregulated processes involved in neuronal development are, e.g., neural tube development at D4, forebrain development at D9, and neurogenesis, nervous system, and neuron projection development at D37. The processes involved in nervous system development are highlighted in blue. A full list of the enriched gene sets can be found in Supplementary Table S1.
Differences in XCI erosion in female cell lines displays little effect on autosomal and X-linked gene expressionSince X chromosome inactivation (XCI) erosion can affect the gene expression of both X-linked and autosomal genes, we investigated the XCI erosion status in our female cell lines. A key indicator for the presence of XCI erosion is the absence of XIST expression. In our study, one of the three primed female cell lines used for RNAseq analysis expresses XIST at a relatively low level (HS980 with 1.5 TPM at D0) without significant changes during differentiation. The other 2 cell lines (WA09/LT2e and HS975) do not express XIST at any time point. To assess the extent of XCI erosion in the female cell lines, we calculated the expression ratio of X-linked genes between male and female cell lines. Since XCI erosion leads to an increased expression of X-linked genes, we can use the XX:XY expression ratio to estimate the degree of XCI erosion. In line with Bar et al., 2019, we consider a ratio of >1.1 as a state in which X-linked genes in female cell lines are increasingly expressed due to XCI erosion. XCI escape genes display an average XX:XY ratio of 1.29 (data not shown) and were removed before the analysis.
On average, the majority of X-linked genes (67%) in the three female lines demonstrate an expression ratio indicative of one X chromosome being inactivated (XaXi), while the remaining 33% of X-linked genes reflect either a state of XCI erosion (XaXe) or two active X chromosomes (XaXa), Supplementary Figure S3A. When analyzed individually, the cell lines exhibit different states of XCI erosion. The lines HS975 and WA09/LT2e display a higher degree of XCI erosion with 31% and 48% of X-linked genes, respectively, with an XX:XY ratio above 1.1 and thus within the XaXe or XaXa range. While in the line HS980 only 16% of X-linked genes display a ratio above 1.1 (Supplementary Figure S3B). To estimate the number of affected genes, we used the average XX:XY gene expression ratio of chromosome 5 and 16 as a baseline, given their similarity in the number of protein-coding genes compared to chromosome X. After subtracting the baseline, the final fraction of X-linked genes upregulated due to the potential effect of XCI erosion was estimated to be on average 6%, corresponding to 38 out of 634 genes (4% or 25 genes for HS980, 4.5% or 29 genes for HS975% and 10% or 63 genes for WA09/LT2e). The complete list of X-linked genes and their corresponding XX:XY expression ratios can be found in Supplementary Table S2. Based on this, only two of the gametologous genes (CASK and SHROOM2) were identified as possibly affected by XCI erosion since they exhibit an XX:XY ratio above 1.1 and are not XCI escape genes. Furthermore, no significant difference was found between the average X-linked gene expression of the cell lines using a one-way ANOVA with Tukey’s multiple comparison test, (Supplementary Figure S3C). Moreover, the variance of the average gene expression from the X-chromosome among the male cell lines (F = 1.33) was twice as high than among the female cell lines (F = 0.65). This suggests that the variation in XIST expression does not critically impact X chromosome gene expression levels in the female lines, rendering them comparable in their X-linked gene expression despite their difference in XIST expression and XCI erosion.
To evaluate whether XIST expression and XCI erosion status affect the autosomal gene expression in the female cell lines, we investigated the relation between X chromosomal and autosomal gene expression. For this, we used the TPM gene expression values to calculate the X:A (Supplementary Figure S3D). We did not observe significant difference in autosomal gene expression in the Xist-positive cell line HS980. A slight trend for increased expression can be noted on chromosomes 8, 9, 11, and 22 with their ratios increased by 0.15–0.2. However, the average X:A ratio of all autosomes was only slightly increased by 0.09 points in HS980, compared to the average of the Xist-negative lines (HS980: 1.16, LT2e: 1.11, HS975: 1.03).
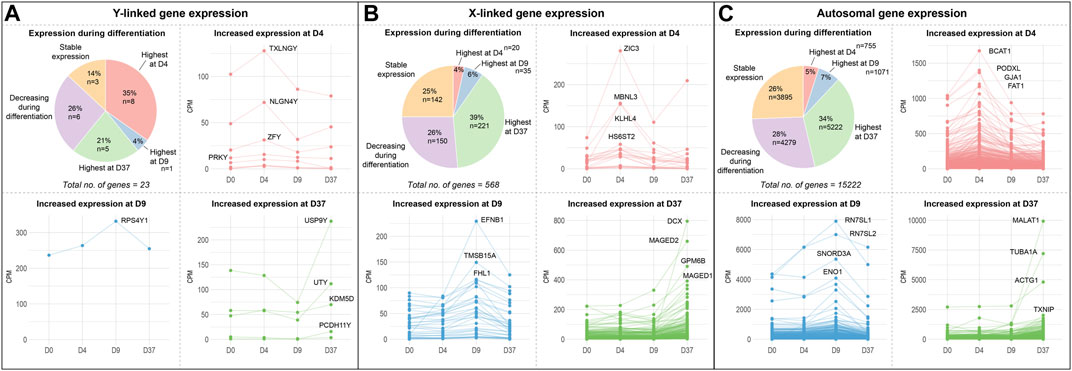
X and Y-linked genes contribute to neuronal differentiationTo identify how many Y-linked, X-linked, and autosomal genes contribute to neuronal differentiation, we investigated their gene expression levels and changes during the differentiation experiment (Figures 2A–C; Supplementary Table S3). We used the average gene expression value of all cell lines grouped by sex and implemented a rigorous threshold by excluding genes with a count number below 50. This stringent criterion was applied to effectively remove a large number of lowly expressed genes. Subsequently, we categorized resulting genes based on the timepoint at which they are expressed highest. We noticed a total of 15,222 autosomal genes, 568 X-linked genes and 23 Y-linked genes to be expressed in the embryonic stem cell lines (Supplementary Table S3). For autosomal and X-linked genes, a similar proportion of genes were upregulated at certain timepoints during differentiation. The largest share of genes was upregulated at D37, accounting for up to 34% of autosomal genes and 39% of the X-linked genes. This is followed by genes that were downregulated during differentiation (26% autosomal, 28% X-linked) and, again in a similar proportion, genes with unchanged expression during differentiation (25% autosomal, 26% X-linked). Only a small percentage of X-linked and autosomal genes seemed to be upregulated at the stages of neuronal induction at D4 (4%–5%) and at D9 (6%–7%). As for the Y-linked genes, the largest number of genes was upregulated early during neuronal induction (35%, 8 genes) at D4. Only one gene is upregulated at D9 in the late stage of neuronal induction (4%, 1 gene).
Figure 2. Gene expression of sex chromosome and autosomal genes during differentiation: Gene expression of (A) Y-linked, (B) X-linked, and (C) autosomal gene expression during neural differentiation. The pie-chart shows the number of genes peaking in expression at different timepoints (D4, D9, or D37), as well as the number of genes decreasing during differentiation and genes that maintained a stable expression level. Additional panels display the expression dynamics of genes peaking at D4, D9, and D37. The genes with the highest expression levels at the corresponding timepoint are labeled. According to their expression peaks, Y-linked genes play a role at different timepoints during neural differentiation in male stem cell lines.
An equal number of Y-linked genes are upregulated at D37 (21%, 5 genes) or decreased during neuronal differentiation (26%, 6 genes), while 14% (3 genes) of Y-linked genes do not change their expression during differentiation. We have labeled the four genes with the highest expression during differentiation at their corresponding timepoints in the panels D4, D9, and D37 in Figure 2. The Y-linked genes with the highest expression were TXLNGY, NLGN4Y, and ZFY at D4, RPS4Y1 at D9, and USP9Y, UTY, KDM5D, and PCDH11Y at D37 (Figure 2A). The X-linked genes with the highest expression were ZIC3, MBNL3, and KLHL4 at D4, EFNB1, TMSB15A, and FHL1 at D9, and DCX, MAGED2, GPM6B, and MAGED1 at D37 (Figure 2B). The autosomal genes with the highest expression were BCAT1, PDOXL, GJA1, and FAT1 at D4, RN7SL1, RN7SL2, SNORD3A, and ENO1 at D9, and MALAT1, TUBA1A, ACTG1, and TXNIP at D37 (Figure 2C). The expression data of these and additional genes, as well as genes that significantly decrease during differentiation or remain stable in expression, can be found in Supplementary Table S3 and Supplementary Figure S4.
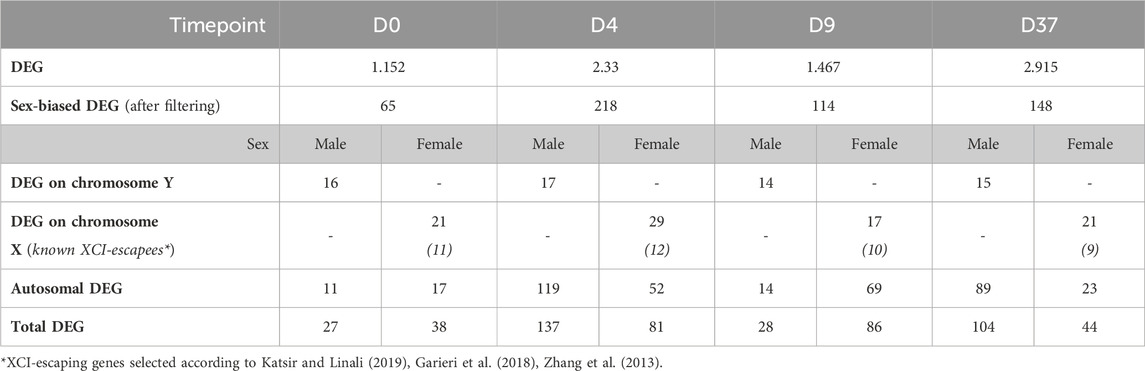
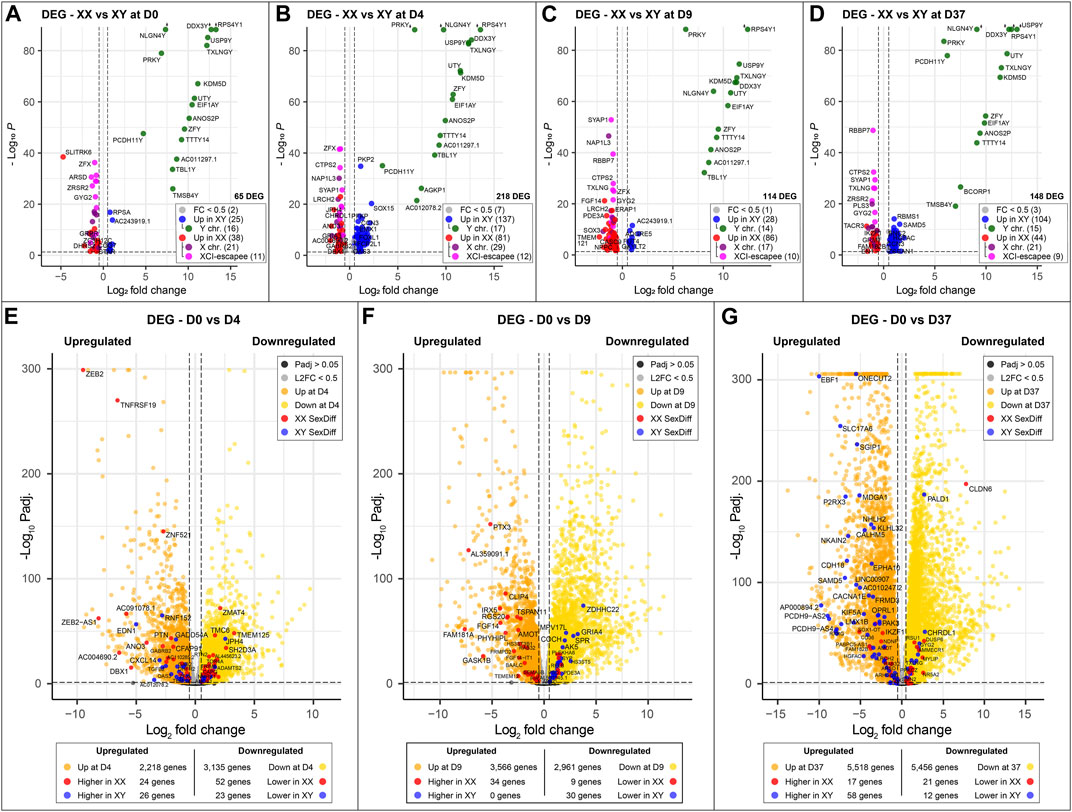
Identifying genes with sex-biased expression and impact on neuronal differentiationTo identify sex-biased gene expression during neuronal differentiation, we performed a differential gene expression analysis with the total RNA sequencing (RNAseq) data. In the assay, we compared 3 male and 3 female cell lines at 4 time points during differentiation, this resulted in the following four comparisons XX to XY at D0, D4, D9, and D37. After applying a filter to exclude genes with low expression (<50 counts), and an adj. p-value >0.05, we obtained 1,152, 2,330, 1,497, and 2,915 differentially expressed genes (DEG) for D0, D4, D9, and D37, respectively (Table 2). However, many of those genes did not meet our requirements for sex-based differential expression. Some DEG displayed a too high variation in expression between the cell lines of one sex to be a reliable candidate for a gene with a sex-biased expression. In other cases, individual cell lines of one sex showed a too similar expression level to cell lines of the opposite sex, so that also these candidates were not a good match for a gene with a sex-bias in expression. To ensure that we select true sex differences, we applied another set of stringent post hoc filtering rules that only assign genes as sex-biased if they are expressed at a similar level within cell lines of the same biological sex (coefficient of variation <0.5), and that required all cell lines of one sex to express a gene at least 20% higher (based on DEseq2 normalized counts) than all cell lines of the other sex. Additional details can be found in the methods section. After filtering, the numbers of differentially expressed genes have reduced substantially to 65, 218, 114, and 148 for D0, D4, D9, and D37, respectively (Table 2). The complete list of DEG can be found in Supplementary Table S4.
Table 2. Number of differentially expressed genes between biological sexes during neuronal differentiation.
As expected, genes of the sex chromosomes (X and Y) make up a considerable part of the sex-biased DEG at all time points (Table 2). The Y chromosome genes display the highest fold-changes among the DEG in male cells. Similarly, the X chromosome genes make up a large part of the overexpressed genes in female cells. Not surprisingly, the X-linked genes that escape X-inactivation (XCI-escapees) show the highest fold-change among the DEG in female cells. Apart from the genes on the sex chromosomes, there is also a significant share of DEG expressed from the autosomes, 43% at D0, 78% at D4, 73% at D9, and 76% at D37. Interestingly, the number of sex-biased DEG increases with the start of differentiation at D4 (Figures 3A–D). While at D0 there are 65 sex-biased genes expressed (27 XY, 38 XX), at D4 it is 218 genes (137 XY, 81 XX), at D9 it is 114 genes (28 XY, 86 XX), and at D37 it is 148 genes (104 XY, 44 XX). Next, we investigated the sex differences at D37 in more detail. Most of the sex-biased genes at D37 are higher expressed in the male cell lines, 104 compared to 44 genes in the female lines (Figure 3D; Supplementary Table S4). From the upregulated genes in the female lines, 21 are X-linked genes, and of these, 9 are known to escape XCI, the remaining 22 genes are autosomal. In contrast, the largest number of upregulated genes in males are expressed from the autosomes (89 genes). Only a small number of the upregulated genes at D37 in the male lines are encoded on the Y chromosome (15 genes). We have annotated all sex-biased genes at D37 for their appearance in neuronal, synaptic, as well as developmental gene sets using the gene ontology database (GO) and collected evidence from available scientific literature regarding their implication in neuronal processes (Supplementary Table S5). Of the 104 DEG in male cell lines, 32 genes were implicated in neuronal processes (Top 5: NLGN4Y, UTY, PCDH11Y, NHLH2, EBF1), while of the 43 DEG in female cell lines it was 7 genes (Top 5: SYAP1, TACR3, GRM1, NDNF, AMOT). Genes that change their expression pattern during neuronal differentiation and do that in a sex-dependent manner are likely to be important candidates that cause sex differences in brain development. To identify these genes, we looked for sex-biased genes among the highly up- and downregulated genes during differentiation (D0 to D4, D0 to D9, and D0 to D37) and found between 2,218 and 5,518 genes that are up- or downregulated, respectively (Figures 3E–G). Approximately 1% of the genes show a sex-bias in gene expression (Supplementary Table S6). We have added this sex-biased expression during neural differentiation as a parameter in Supplementary Table S5 and used it for the selection of candidate genes later.
Figure 3. Sex-specific differences at key timepoints and in genes that change significantly during differentiation: (A–D) Sexually differentially expressed genes comparing male (XY) against female (XX) cell lines at different time points of neuronal differentiation. Genes overexpressed in male cell lines are marked in blue; green if they are located on the Y chromosome. Similarly, genes overexpressed in female cell lines are marked in red; purple if they are expressed from the X chromosome and pink if they escape XCI. The number of sex-biased genes overexpressed in male and female cell lines is as follows: (A) D0, 65 genes in total, 25 in XY and 38 in XX; (B) 218 genes in total, 137 in XY and 81 in XX; (C) 114 genes in total, 28 in XY and 86 in XX; (D) 148 genes in total, 104 in XY and 44 in XX. The number of genes with sex-biased expression from sex chromosomes remains relatively constant with an average of 15 Y chromosome and 22 X chromosome genes expressed at each time point. XCI-escapee genes are among the sex-biased genes with the highest p-value in the XX cell lines. (E, F) display the DEG that are up- and downregulated over the course of neuronal differentiation. Genes that are additionally expressed in a sex-biased manner are highlighted in red and blue according to their sex-bias in female and male cell lines, respectively. On average, 3,500 genes are up- and downregulated at each time point, of which 1% display sex-biased expression. (E) From D0 to D4: 2,218 genes are upregulated (26 in XY and 24 in XX with a sex-bias) and 3,135 are downregulated (23 in XY and 52 in XX with a sex-bias); (F) From D0 to D9: 3,566 genes are upregulated (0 in XY and 34 in XX with a sex-bias) and 2,961 are downregulated (30 in XY and 9 in XX with a sex-bias); (G) From D0 to D37: 5,518 are upregulated (58 in XY and 17 in XX with a sex-bias) and 5,456 are downregulated (12 in XY and 21 in XX with a sex-bias).
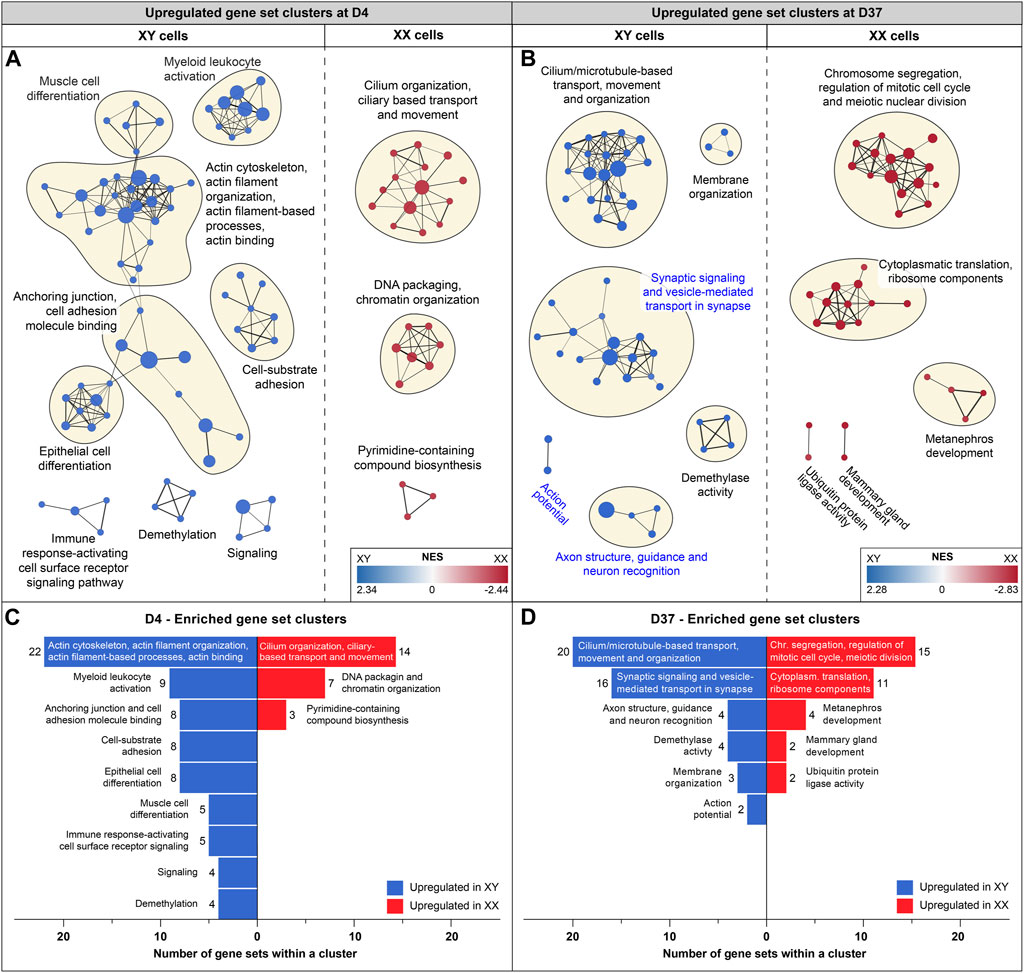
Sex-biased gene expression in neural processes after 37 days of differentiationTo identify potential differences in biological processes between cell lines of different sexes, we conducted additional GSEAs comparing male and female gene expression at the timepoints D0, D4, D9, and D37. Enrichment maps were generated from the GSEA results to identify clusters of upregulated gene sets with similar functions. Clusters of gene sets are more likely to have a biological relevance than isolated gene sets. Using this approach, identified various biological processes before differentiation and at the early (D4) and late stages of differentiation (D37). Interestingly, at D9, the analysis did not reveal any differences between the cell lines. Before differentiation (D0), upregulated gene set clusters in male cell lines were associated with ribosomal and mitochondrial functions (Supplementary Figure S5). At D4, a substantial number of actin-based processes were increased in male cells, while in female cells, cilium-associated processes, as well as DNA packaging and chromatin organization, were upregulated (Figures 4A, C). At D37, male cells exhibited upregulation in cilium and microtubule-based processes, as well as in various neural processes such as synaptic signaling, synaptic vesicle transport, axon structure, axon guidance, neuron recognition, and action potential. In contrast, female cell lines were upregulated in cell division and ribosomal processes (Figures 4B, D). Details for all enriched gene sets within a cluster can be found in Supplementary Table S7.
Figure 4. Enriched gene sets at D4 and D37: Gene set enrichment analysis was based on differentially expressed genes at D4 and D37. Enriched gene sets in male and female cell lines are colored in blue and red, respectively. Panels (A, B) display an enrichment map in which a dot represents an upregulated gene set. If gene sets are connected, and the closer they are in proximity, the more they are related in function. Clusters of gene sets are encircled and named after their common biological function. (A) Enrichment map for D4: male cell lines display upregulated gene sets that are associated with actin-based processes, such as “Cell-substrate adhesion”, “Anchoring junction” and “Actin cytoskeleton and actin filament organization”. Female cell lines at D4 display larger gene set clusters of cilium-based processes as well as “DNA packaging and chromatin organization”. (B) Enrichment map for D37: male cell lines show three clusters of enriched gene sets that are associated with neuronal processes (cluster names highlighted in blue) and a larger cluster that combines cilium and microtubule-based processes. Female cell lines at D37 show an upregulation of gene sets involved in cell division and ribosomal processes. Subfigures (C) and (D) summarize upregulated gene sets in male and female cell lines (at D4 and D37) and display the number of gene sets within a cluster.
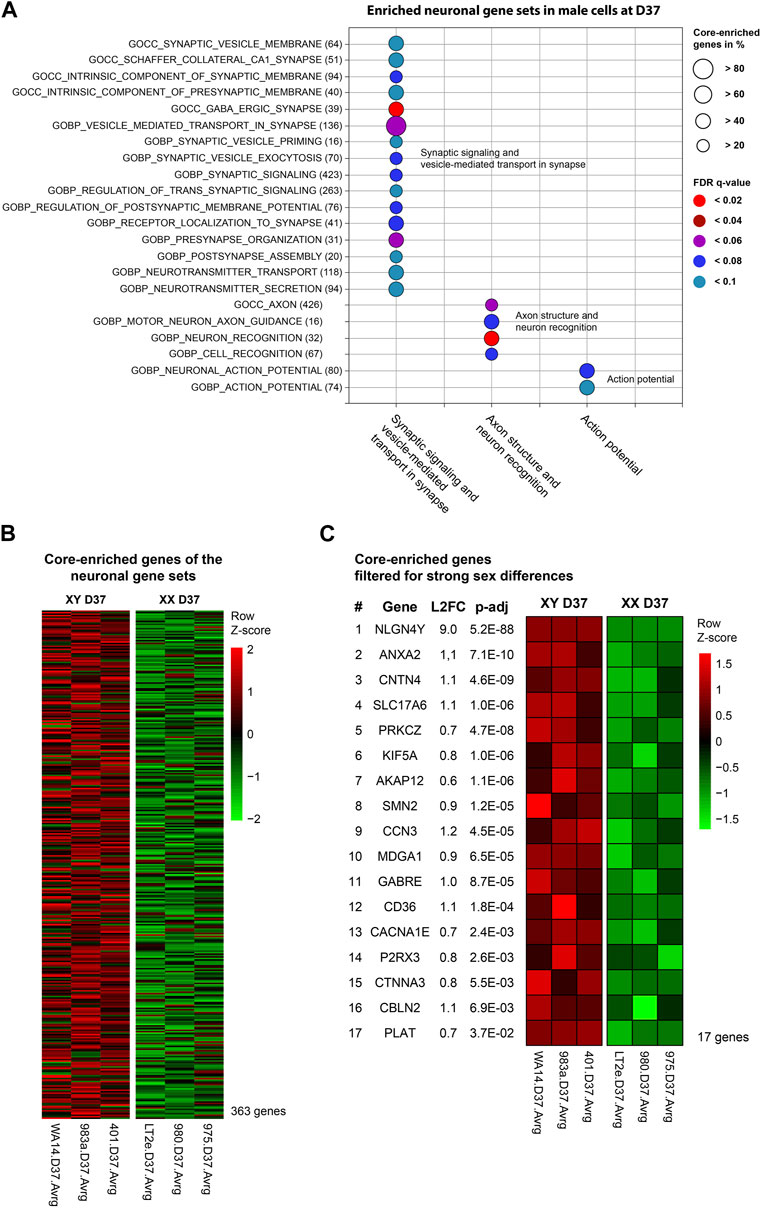
At D37, the GSEA identified three clusters of sex-biased neural processes in male cell lines. To pinpoint the specific genes contributing to these clusters, consisting of 22 individual gene sets, we extracted the genes with the highest contribution to the gene set enrichment, known as core-enriched genes (Figure 5A). This resulted in 362 unique core-enriched genes (Figure 5B), which we further filtered using previously mentioned stringent filtering rules, to identify 17 sex-biased genes implicated in neural processes (Figure 5C). The full list of core-enriched genes is available in Supplementary Table S8, with the 17 sex-biased genes highlighted.
Figure 5. Enriched neuronal gene sets and corresponding core-enriched genes in male cell lines at D37: Core-enriched genes from enriched neuronal gene sets in male lines at D37 were contributing to candidate gene selection. (A) Gene sets upregulated within the neuronal clusters. Numbers in brackets show the total number of genes within the gene set. Circle size illustrates the percentage of core-enriched (highly contributing) genes within the gene sets. Color indicates the false-discovery rate (q-value) of the gene set at comparison of male and female cell lines. (B) Gene expression heatmap of core-enriched genes from all gene sets within the sex-biased neural process clusters. Colors indicate the level of expression comparing male and female cell lines based on a Z-score. Red indicates an overexpression, black indicates an equal expression in both cell lines, green indicates a reduced expression. (C) Using stringent filtering, we identified 17 core-enriched and significantly sex-biased genes within the clusters. These genes later contributed to the selection of candidate genes.
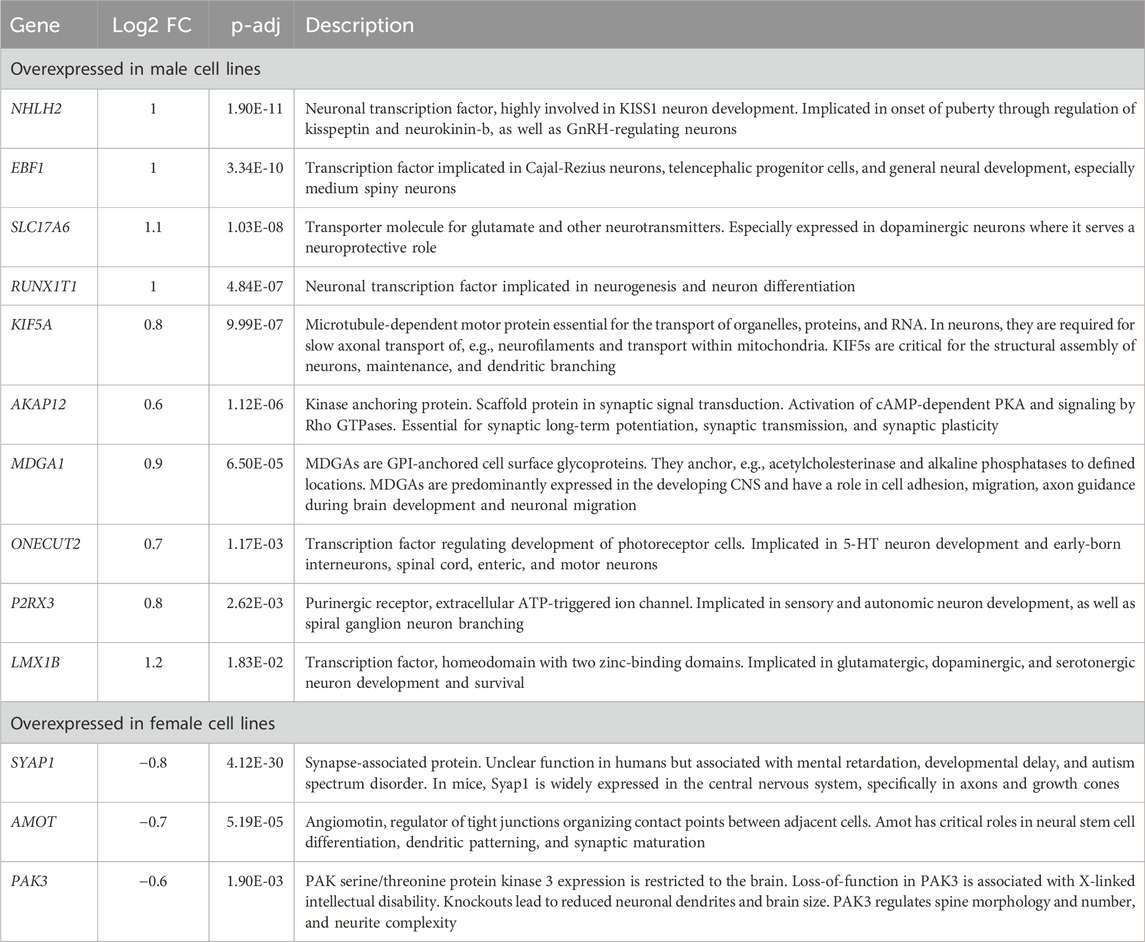
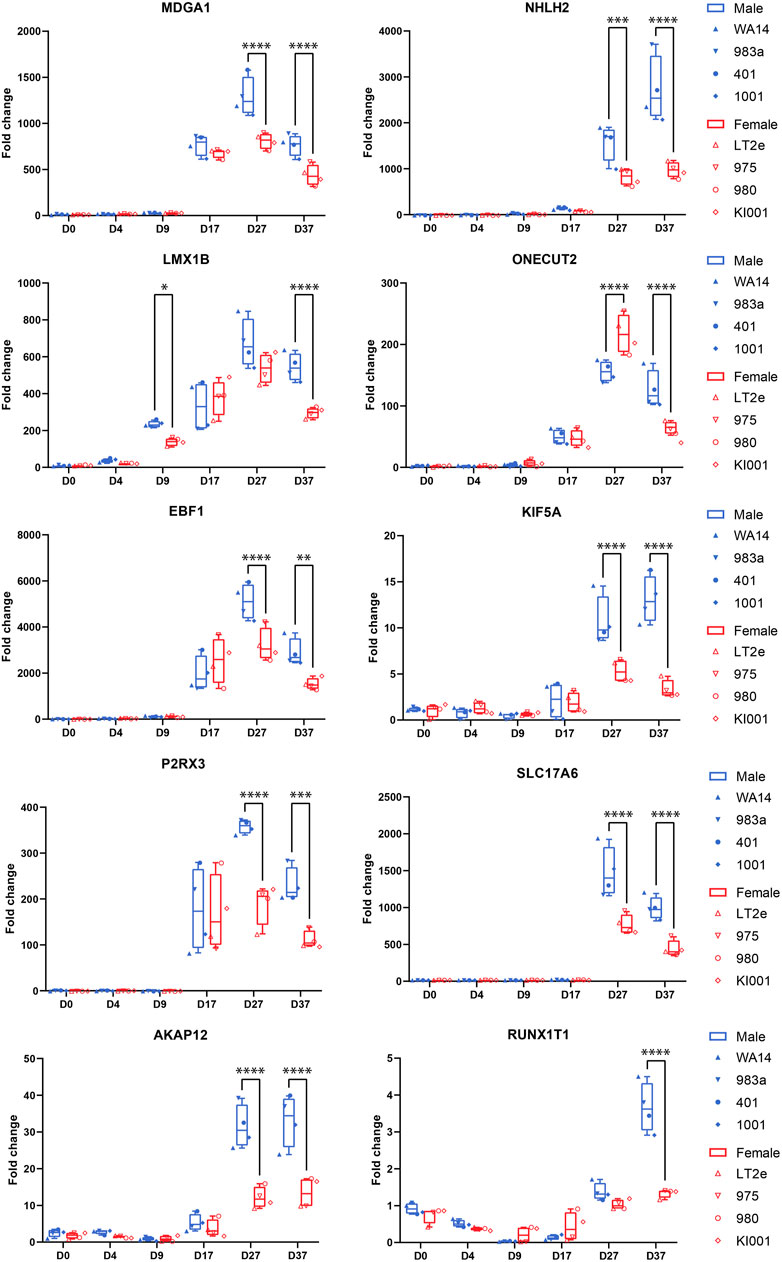
Combined information returns candidate genes that show robust sex-biased overexpression in the cell lines and are implicated in neural processesNext, we aimed to identify candidate genes demonstrating robust sex differences in expression and implication in the development and function of neurons. For this, we combined all the information gathered in this study to annotate the sex-biased genes that resulted from the DEG analysis at D37 (104 upregulated genes in XY cells and 44 in XX cells). The following information was used for the annotation: (a) GO-terms associated with neurodevelopment, (b) upregulation at D37 of differentiation, (c) appearance in clusters of neuronal processes in the GSEA, (d) indications from the literature for involvement in neurodevelopmental processes, and (e) the 17 sex-biased genes resulting from the GSEA analysis of D37. Based on these annotations more than 60 of the 148 genes display implications in neuronal development. Among these genes, 13 displayed an expression level above the 70th percentile, which we decided to select as candidate genes. Ten of these genes display an upregulated gene expression in male cell lines (NHLH2, EBF1, SLC17A6, RUNX1T1, KIF5A, AKAP12, MDGA1, ONECUT2, P2RX3, LMX1B), and 3 genes are upregulated in female cell lines (SYAP1, AMOT, PAK3), as detailed in Table 3. The full list of annotated genes can be found in Supplementary Table S5. To validate the expression tendencies of the candidate genes, we conducted qPCR analyses on material from a replicate differentiation experiment, including two additional cell lines for each sex (KARO1-XX and HS1001-XY) and two extra timepoints, D17 and D27. By including timepoints D17 and D27, we observed that most candidate genes display an initial expression earliest at D17 (Figure 6). Considering this and the onset of DCX expression at D17 (Supplementary Figure S2), we regard D17 as the earliest timepoint where neurons begin to emerge in our differentiation experiment. The sex-biased expression of candidate genes was affirmed at D37 in all cell lines, with some genes even exhibiting a sex difference as early as D27. The difference between the male and female samples was tested using a repeated measurements two-way ANOVA with Šidák’s multiple comparisons test.
Table 3. Description of candidate genes: Sex-biased genes overexpressed in male (pos. Log2FC) or female cell lines (neg. Log2FC) with implications in neuron development.
Figure 6. Candidate gene expression at all time points: Gene expression was measured by qPCR in replicates of the neural differentiation experiment (n = 3). All candidate genes (MDGA1, NHLH2, LMX1B, ONECUT2, EBF1, KIF5A, P2RX3, AKAP12, SLC17A6, RUNX1T1) show significant overexpression in all four male cell lines at D37. Many genes start expressing at D17. RUNX1T1 shows a significant sex difference already at D9. ONECUT2 shows a female-biased sex difference at D27, which then changes to an overexpression in male cells at D37. Differences between the male and female samples were measured using repeated measurements two-way ANOVA with Šidák’s multiple comparisons test.
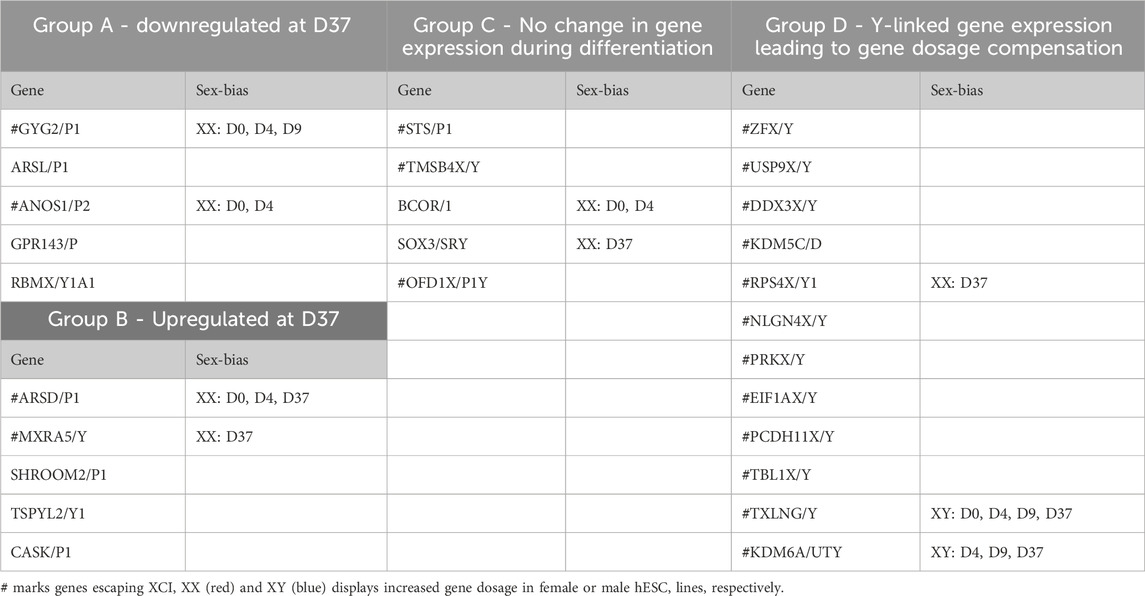
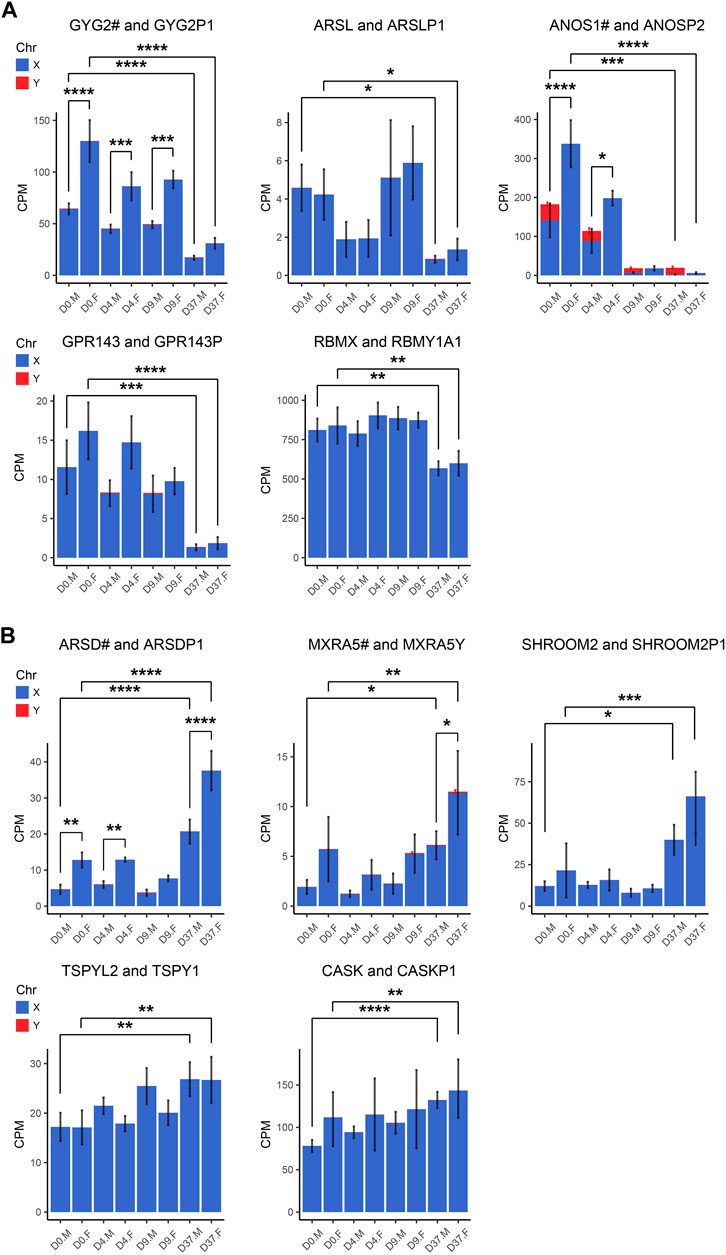
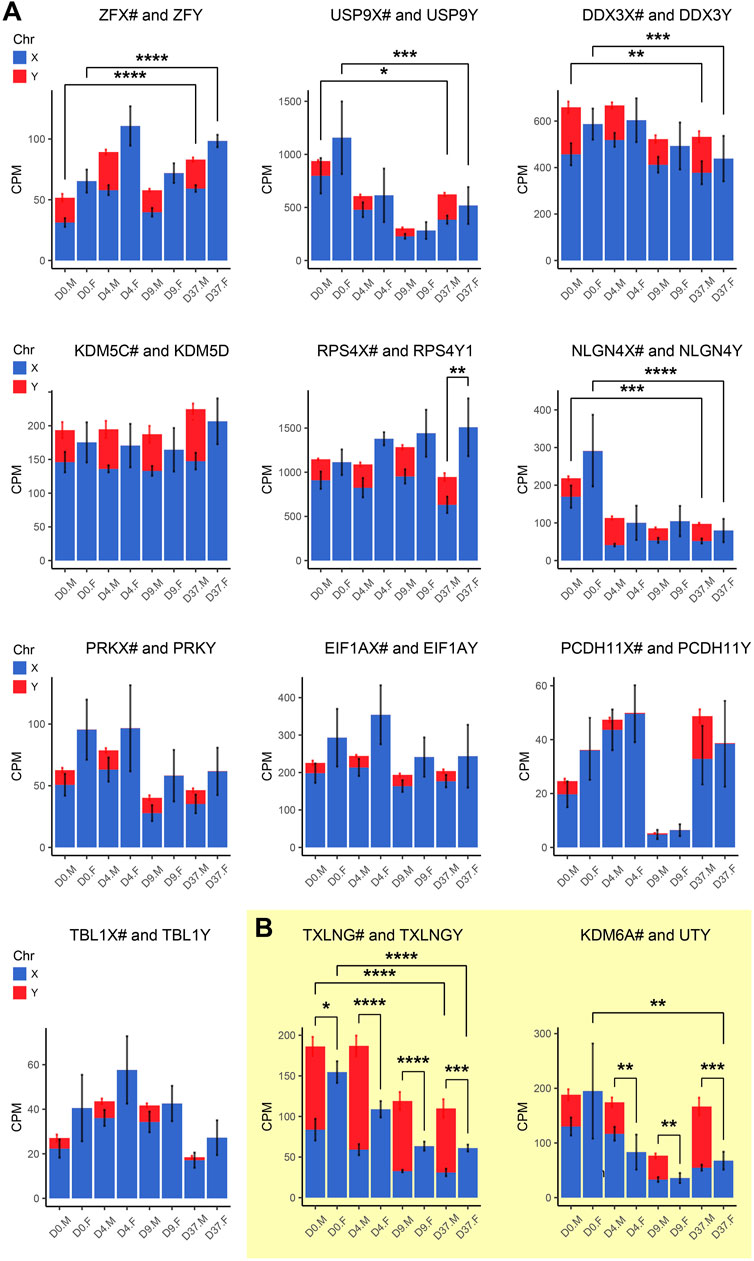
Gene dosage compensation of X/Y homologs escaping XCI through Y homolog and significant Y homolog overexpression in TXLNG/Y and KDM6A/UTYGametologous genes are X/Y homologous genes located on opposite sex chromosomes in non-recombining regions. Gene dosage of gametologous genes can contribute to sex-biased gene expression. The analysis of these genes in male and female cell lines has revealed several sex differences (Table 4). Since the impact of XCI erosion on the gene expression of X-linked genes cannot be entirely ruled out, the interpretation of gametologous gene expression should be done with caution, particularly when there is a significantly increase of expression in the X homologs in this section. We categorized the gametologous gene pairs into four groups based on their expression pattern during the neural differentiation period from D0 to D37. Group A includes genes that are significantly downregulated at D37 (GYG2/P1, ARSL/P1, ANOS1/P2, GPR143/P, RBMX/Y1A1; Figure 7A). In contrast, Group B comprises genes upregulated at D37 (ARDS/P1, MXRA5/Y, SHROOM2/P1, TSPYL2/TSPY, CASK/P1; Figure 7B), while Group C shows no change in gene expression from D0 to D37 (STS/P1, TMSB4X/Y, BCOR/1, SOX3/SRY, OFD1X/P1Y; Supplementary Figure S6). Group D includes all gene pairs that show substantial Y chromosome homolog expression contributing to gene dosage compensation (ZFX/Y, USP9X/Y, DDX3X/Y, KDM5C/D, RPS4X/Y1, NLGN4X/Y, PRKX/Y, EIF1AX/Y, PCDH11X/Y, TLX1X/Y; Figure 8A). This group includes two gene pairs (TXLNG/Y, KDM6A/UTY) that display a significant overexpression of the Y chromosome homolog leading to sex biases in gene dosage, Figure 8B. More than half of the X-linked genes of the X/Y homologous gene pairs (19 out of 27) are escaping XCI according to Katsir and Linial (2019), Garieri et al. (2018) and Zhang et al. (2013). We observe that for many of the XCI-escapees (12 out of 19), the Y chromosome compensates the increased X chromosome expression, and therefore, these gene pairs do not contribute to sex differences. These genes were clustered in Group D, with the exception of one gene (ANOS1), which sharply drops in gene expression at D37 and thus is categorized into Group A.
Table 4. Grouping of X/Y homologs according their expression during neuronal differentiation and sex-bias in gene dosage.
Figure 7. Gametologous genes with low Y homolog expression: X/Y homolog expression was analyzed in total RNA sequencing data (3 male and 3 female cell lines). A hashtag after the X homolog indicates that the gene is escaping XCI. Panel (A) displays the 5 gene pairs that decrease (GYG2/P1, ARSL/P1, ANOS1/P2, GPR143/P, RBMX/Y1A1), and (B) the 5 gene pairs that increase (ARDS/P1, MXRA5/Y, SHROOM2/P1, TSPYL2/TSPY, CASK/P1) in expression during neural differentiation, comparing the period D0 to D37. An increased gene dosage in females was found in GYG2/GYG2P1 at D0, D4, and D9 (p < 0.0001, p < 0.001, p < 0.001, respectively) and ANOS1 at D0 and D4 (p < 0.001, p < 0.05). Differences between the male and female samples were measured using repeated measurements two-way ANOVA with Šidák’s multiple comparisons test.
Figure 8. Gametologous genes with high Y homolog expression: Gene expression was analyzed based on total RNA sequencing data. The hashtag symbol after the X homolog indicates that the gene is escaping XCI. Panel (A) shows gene expression of 10 gametolog pairs that show substantial Y chromosome homolog contribution to gene dosage balance (ZFX/Y, USP9X/Y, DDX3X/Y, KDM5C/D, RPS4X/Y1, NLGN4X/Y, PRKX/Y, EIF1AX/Y, PDCDH11X/Y, TLX1X/Y). A sex difference was noticed in RPS4X/Y, with an increased gene dosage in female cell lines (p < 0.01). Panel (B) displays two gene pairs (TXLNG/Y, KDM6A/UTY) that display excessive Y chromosome homolog expression leading to an unbalanced gene dosage. For TXLNG/Y, this led to a male bias in gene dosage at all time points D0, D4, D9, and D37 (p < 0.05, p < 0.0001, p < 0.0001, p < 0.001, respectively). For the gene pair KDM6A/UTY, a male-biased gene dosage was found in D4, D9, and D37 (p < 0.01, p < 0.01, p < 0.001, respectively). Differences between the male and female samples were measured using repeated measurements two-way ANOVA with Šidák’s multiple comparisons test.
We detected a female-biased overexpression of gametologous genes in Group A (GYG2, ANOS1, BCOR1) during early differentiation (D0, D4, D9). This sex bias could contribute to different prerequisites that affect subsequent differentiation steps. We also noticed a female-biased overexpression in genes of Group B: ARSD at D0, D4, and D37 and MXRA5 at D37. A sex bias in Group B at D37 suggests relevance in late neural differentiation or maintenance. Of the Group C genes, the gene RPS4X is significantly overexpressed in females at D37 due to insufficient Y chromosome gene dosage compensation. Moreover, the Y homolog expression of the Group D gene pairs TXLNG/TXLNGY and KDM6A/UTY is very high at nearly all timepoints, leading to a significant increased gene dosage in male cell lines.
DiscussionUnderstanding sex differences in the field of neuroscience is important to predict, monitor and treat neurological disorders, especially since a majority of early-onset neurodevelopmental disorders show a clear sex bias (Boyle et al., 2011; Loke et al., 2015; Tesic et al., 2019). In this study, we sought to determine sex-specific gene expression during early neural differentiation. We used four male and four female-derived hESC lines and applied a differentiation protocol resulting in a population of mixed neurons. Using hESCs as a research model has several advantages. By using epiblast-derived embryonic stem cells, we aimed to keep the epigenetic variability low, that can otherwise arise as a result of “the epigenetic memory” of the cell-of-origin when cells are reprogrammed to an induced pluripotent state (Narsinh et al., 2011). Furthermore, we can exclude the effects of sex hormones by using a cell culture model in a controlled in vitro environment. Thus, we consider our model a valuable system to study genetic sex differences that affect early nervous system development. It proved to be a robust system as it resulted in reproducible and comparable differentiation in all cell lines (Figures 1B–E). While our focus was primarily on neurons, it is important to acknowledge the complexity of neural development, which encompasses various cell types such as astrocytes, oligodendrocytes and microglia. Our findings demonstrate that under the culture conditions employed, neuronal development was favoured, with no discernible markers for other cell types within the neuronal lineage detected. While it's important to interpret these findings thoughtfully, it's worth noting that the culture conditions utilized, though not exact replicas of organotypic environments, still provide valuable insights contributing significantly to our understanding of sex-specific mechanisms in neuronal differentiation.
While X-linked genes have already been extensively linked to the development and functions of the nervous system (Skuse, 2005; Nguyen and Disteche, 2006; Mallard et al., 2021; Raznahan and Disteche, 2021; Cabrera Zapata et al., 2022), evidence for the involvement of Y-linked genes in male neuronal development is scarce (Vakilian et al., 2015; Meyfour et al., 2017; Johansson et al., 2019; Pottmeier et al., 2020; Cabrera Zapata et al., 2022). In this study, we confirm the involvement of X- and Y-linked genes in the neuronal differentiation of male and female human embryonic stem cells. Furthermore, we provide evidence for sex-biased gene expression during neural differentiation, most prominent after 37 days of differentiation, and highlight 13 candidate genes that show sex-biased expression, thus possibly impacting neuronal development. Finally, we display gene dosage compensation of X homologs that escape X-inactivation through their Y homolog in male cell lines suggesting a more important role for the Y chromosome in neurodevelopment than generally believed.
The comparison of gene expression in male and female stem cell lines presents a number of challenges. One of them, which specifically affects X-linked gene expression is the state of XCI in female stem cell lines. It is well-documented that commonly used cell culture conditions lead to the erosion of XCI in female cell lines (Hanna et al., 2010; Cloutier et al., 2022), which in turn can result in increased expression of X-linked and autosomal genes. The absence of XIST expression in two of the female cell lines and the number of genes with an XX:XY expression ratio above 1.1 indicates different XCI erosion statuses in our cell lines. However, despite this erosion, the very similar expression of X-linked genes and the low number of X chromosome genes with an XX:XY expression ratio above 1.1 indicates that the XCI erosion has only a minor effect on their X-linked gene expression. The same is true for the autosomal gene expression as the comparison of the average X:A ratio revealed no significant increase in the XIST positive cell line. In addition, our female cell lines were able to differentiate equally well to neurons, whereas under XCI erosion cell lines have been reported to be impaired in neural differentiation (Motosugi et al., 2022). Moreover, we must consider whether our analyses to detect sex differences in the study might be influenced by genes exhibiting high variance, possibly attributed to the erosion of XCI. Here, it's worth noting that such genes were excluded based on the stringent filtering criteria we implemented for identifying sex-biased genes. Consequently, we assert that the findings of our study remain unaffected by the varying XCI erosion statuses observed in the XX cell lines.
The use of the XX:XY ratio to estimate the number of genes affected by XCI erosion is an approximation and may not cover all cases. The observed disparities for chromosomes X, 5, and 16 reflect a combination of cell-line-specific variability and authentic effects of XCI erosion, which are inherently intertwined and indistinguishable. Certain scenarios may remain unaccounted for, such as instances where physiologically low X-linked gene expression in a female cell line, yielding an XX:XY ratio of 0.5, is subsequently doubled due to the reactivation of the second X chromosome through XCI erosion. In this scenario, although the gene is affected, it does not meet the criteria for being considered “affected by XCI erosion” (ratio>1.1). This methodology assumes uniform X-linked gene expression between female and male cells under identical experimental conditions. We consider this approach suitable to estimate the general progression of XCI erosion and its impact on X-linked gene expression in female cell lines, given that average X-linked expression levels are commonly reported to be elevated in XIST–hPSCs compared to XIST+ and male hPSCs (Bar et al., 2019; Motosugi et al., 2022; Sarel-Gallily and Benvenisty, 2022). Nevertheless, results including X-linked genes should be interpreted with caution as a possible effect from XCI erosion cannot be entirely excluded.
Considering that the majority of gametologous genes are XCI escape genes (19 out of 27), transcribed from two X chromosomes, it is improbable that XCI erosion significantly affects their gene expression. Only two gametologous genes (CASK and SHROOM2) exhibit signs of XCI erosion, as indicated by an XX:XY ratio above 1.1 and are not XCI escape genes. Therefore, we consider the analysis of gametologous genes presented in this study to be largely unaffected by XCI erosion.
Genetic sex differences are not routinely investigatedEven though ‘sex differences’ is a subject that receives increasing awareness among researchers and is regularly debated among neuroscientists (DeCasien et al., 2022), the investigation of sex-biased gene expression is not pursued frequently. Most studies that investigate sex-biased gene expression during neurodevelopment are transcriptomic studies that utilize brain tissues of the developing embryo (Shi et al., 2016; O’Brien et al., 2018; Johansson et al., 2019; Liu et al., 2020; Oli
留言 (0)