Animals
Male C57BL/6 mice, aged 8–10 weeks and weighing between 25-30 g, were purchased from Zhejiang Vital River Laboratory Animal Co., Ltd. The mice were kept in a controlled environment, with a regulated temperature and humidity, and a 12-h light/dark cycle. All animal experiments described in this study were carried out in compliance with the Animal Care and Use Committee regulations.
Experimental design
I experimental design was depicted in Additional file 1: Part S1.
ICH model
ICH model was established by injection of collagenase into mice striatum [30]. Briefly, a 0.3% solution of pentobarbital sodium was utilized for anesthesia. Prior to injection, the mice were weighed and then received an intraperitoneal injection of 0.1–0.2 ml/10 g of the anesthetic agent. After complete anesthesia, mice were fixed onto the brain stereotaxic frame, and the head retainer was adjusted to maintain the horizontal position of the mouse’s head. The scalp hair was shaved, and the surgical area was cleaned with an alcohol swab. A 0.5–1 cm longitudinal incision was made to expose the skull, and the anterior fontanelle was uncovered. The needle of a Hamilton syringe was then inserted through a burr hole into the right basal ganglia at coordinates: 0.5 mm anterior, 2.2 mm lateral, and 3.5 mm ventral to the bregma. After confirming the injection site, the skull was opened with a grinding drill, and 0.5 μl of normal saline solution containing 0.0375U collagenase was injected into the right striatum of the mice at a uniform rate of 0.1 μl/min using a microinjection pump. Once the injection was completed, the microsyringe remained in situ for 5 min before being slowly removed. The burr hole was sealed with bone wax, and skin incision was then closed. The operation procedure was identical in the sham operation group, except no collagenase was administered. The operation was performed under sterile conditions, and the mice’s body temperature was maintained at 37 ℃ using a heating pad. Following the operation, mice were resuscitated with 0.4 ml sterile saline, and their body temperature was sustained. Mice were monitored closely until fully recovered from anesthesia. Once resuscitated, mice had free access to food and water.
Drug administration
According to the previous research literature, intranasal administration can improve the central utilization rate and reduce the peripheral side effects by delivering the drug to the brain via the olfactory nerve and ethmoid sinus, and it can be administered repeatedly for a long time [31]. In order to facilitate clinical transformation, this study was conducted by intranasal administration. AVE0991 at dosages of 0.3, 0.9, and 2.7 mg/kg dissolved in 10% dimethyl sulfoxide (DMSO) mixed with corn oil and intranasally administered 1 h after ICH for 3 consecutive days as previously reported [23]. A779 (3 nmol in 2 µL), a selective inhibitor of Mas receptor, was dissolved in PBS and administered intracerebroventricularly (i.c.v.) 1 h before ICH injury. Intracerebroventricular administration was performed as previously described [32]. Intracerebroventricular injection was executed at a flow rate of 0.25 μL/min with the aid of the following coordinates referenced from bregma: 0.3 mm posterior, 1 mm right lateral, and 2.3 mm ventral. ML385 (30 mg/kg), a selective inhibitor of Nrf2, was dissolved in 5%DMSO and administered intraperitoneally at 1 h prior to ICH induction.
Neurobehavioral tests
In this study, the modified neurological deficit score (mNSS), corner turning test, and forelimb placement test (FP test) were utilized to assess the short-term neurofunctional behavior of mice (1, 3, 5, and 7 days after ICH) [32, 33]. The medium- and long-term neurofunctional behavior of mice was evaluated through the open field test (14 days after ICH) and the Morris water maze test (23–28 days after ICH).
The overall score of the mNSS was 18, which was determined by assessing movement, sensation, balance, and reflexes in mice. The severity of symptoms was directly proportional to the score obtained. In the corner turning test, mice were observed over 10 trials and a score was determined by calculating the percentage of right turns out of 10 trials. For the forelimb placement test, the positioning of the left forelimb on the countertop was recorded upon stimulation of the vibrissae. The percentage of times the left forelimb was positioned correctly out of ten consecutive vibrissae stimulations was then calculated.
For the open field test, experimental mice were randomly placed at the center of the test box and recorded video for 10 min. After each experiment, the total distance moved and central area distance moved by the mice were recorded and calculated within the 10-min timeframe.
The Morris water maze test was performed on days 23–28 after ICH to evaluate the spatial learning and memory capabilities, which has been previously reported [32, 34]. The water maze device consisted of a circular cylindrical pool that was divided into four quadrants and equipped with an image acquisition system. The water in the pool was colored milky white, and a 10 cm platform with a diameter ranging from 0.5 to 1 cm was situated in one of the quadrants, with azimuth marks positioned around the pool to serve as cues for the mice. The experimental mice were allowed to swim for up to 60 s, starting at different quadrants in each trial and stayed on the platform for 15 s. On day 28, the platform was removed, and the mice were placed into the pool from the opposite side of the original platform quadrant. The complete movement tracks of the mice within 60 s were recorded, and the percentage of stay time and the number of platform crossings in the quadrant of the original platform were subjected to statistical analysis.
Liquid Chromatograph-Mass Spectrometer (LC–MS) analysis
The brain samples were prepared as previously described [23]. For LC–MS analysis, the entire brain was weighed and mixed with 1.3 mL of methanol. After grinding the tissue for 5 min and gently scrolling for 5 min more, the sample was transferred to a 1.5 mL centrifuge tube and subjected to 10 min of centrifugation at 13,000 rpm. The resulting supernatant was carefully collected and filtered using a 0.22 μm filter membrane before being injected into the LC–MS system. The mass spectrometer (ThermoFisher) was set to the positive ion scanning mode and selective reaction monitoring (SRM), while the chromatograph (ThermoFisher) adopted the Welch Ultimate XB-C8 150 4.6 mm, 5 μm column. The chromatogram was collected and analyzed using Xcalibur software (ThermoFisher).
Hematoma volume and hemoglobin content measurement
The measurement of hematoma volume was conducted in accordance with established protocols as previously reported [32]. This assessment was performed at intervals of 3 and 7 days after inducing ICH. In brief, the mice were placed under deep anesthesia and transcardially perfused with 4 °C phosphate-buffered saline (PBS). The entire brain was removed and sliced coronally at 1-mm intervals, with each section photographed. The hematoma volume was calculated using the formula: hematoma volume (mm3) = area of each section (mm2) × number of sections × thickness (mm), as determined using the Image J software.
Hemoglobin assay was carried out using Drabkin's reagent to quantify hematoma according to previously published methods [32]. Following homogenization and ultrasonic lysis of the brain sections, the samples were centrifuged to obtain the supernatant. 0.4 mL of Drabkin’s reagent (Sigma-Aldrich) was added to 0.1 mL of supernatant and allowed to react for 15 min at room temperature. The hemoglobin absorbance was measured by a spectrophotometer (ThermoFisher) at 540 nm. The data was then calculated as a ratio to the sham group, with the preset value of 1 assigned to the sham group [35].
Brain water content measurement
After inducing deep anesthesia, the mice were quickly decapitated, and the entire brain tissues were immediately taken out. The olfactory bulb was removed. The brain tissue was then divided into left and right hemispheres as well as the cerebellum, which were weighed using an electronic balance and recorded as wet weight. Subsequently, the three portions were placed in a constant temperature oven at 100 ℃ for 3 days before being weighed again and recorded as dry weight. The brain water content was calculated as (wet weight-dry weight)/wet weight*100%.
Enzyme‑linked Immunosorbent Assay (ELISA)
Serum and brain tissue levels of Ang-(1–7) were quantified using a commercially available Mouse Angiotensin 1–7 ELISA kit (CUSABIO) in accordance with the manufacturer's instructions. Each ELISA analysis was performed in triplicate. To quantify Ang-(1–7) levels, the absorbance of samples was read and analyzed at 450 nm using a spectrophotometric plate reader (ThermoFisher).
Quantitative real-time polymerase chain reaction (q-PCR)
The surrounding brain tissues of the hematoma were used for q-PCR analysis. Total RNA was extracted using the RNA extraction reagent (TRIzol, Invitrogen) following the manufacturer's instructions. Subsequently, cDNA synthesis and subsequent qPCR were performed using the Servicebio® RT First Strand cDNA Synthesis Kit (Servicebio) and SYBR Green qPCR Master Mix (without ROX, Servicebio). The synthetic primer sequences are listed in Additional file 1: Part S2.
Western Blot analysis
Under deep anesthesia, mice were transcardially perfused with cold PBS, and brains were extracted and surrounding tissues of the hematoma were dissected. Total proteins of brain tissues were extracted using SD-001/SN-002 Minute™ Total Protein Extraction Kit (Invent Biotechnologies). Equal amounts of protein were loaded on an SDS-PAGE gel and run using electrophoresis and then transferred to a nitrocellulose membrane. The membrane was blocked with 5% nonfat blocking grade milk and then incubated overnight at 4 °C with the following primary antibodies: rabbit anti-MPO (1:1000, ab208670, Abcam), rabbit anti-IL-1β (1:1000, 12,426, Cell Signaling Technology), rabbit anti-Bax (1:5000, 50,599–2-Ig, Proteintech), rabbit anti-Bcl-2 (1:2000, 26,593–1-AP, Proteintech), rabbit anti-p-Akt (Ser473, 1:1000, #4060, Cell Signaling Technology), rabbit anti-Akt (1:10,000, 60,203–2-Ig, Proteintech), rabbit anti-Nrf2 (1:1000, 12,721, Cell Signaling Technology, USA), rabbit anti-iNOS (1:1000, ab178945, Abcam), rabbit anti-CD163 (1:1000, 16,646–1-AP, Proteintech). The membranes were incubated with a rabbit monoclonal antibody anti-β-actin (1:20,000, AC026, ABclonal) as an internal reference. The appropriate secondary antibodies (1:5000, Cell Signaling Technology) were chosen for 2 h of incubation at room temperature. The density of the bands was measured by ImageJ software (NIH) and expressed as a ratio between the intensity of the target band and that of β-actin band.
Immunofuorescence staining
After being anesthetized deeply, mice were transcardially perfused with cold PBS and 4% paraformaldehyde. Then the brains were removed and placed successively in 4% paraformaldehyde, 20% sucrose, and 30% sucrose for complete fixation and dehydration. The embedded brain tissues were sliced along the coronal plane with a thickness of 30 μm using a constant temperature frozen slicer. After being washed in PBS and PBS + 0.3% Triton, coronal sections were incubated in PBS + 1% Triton to break the cell membrane and blocked with immunofluorescence blocking solution for 1 h. The brain sections were then incubated overnight at 4 °C with primary antibodies including: rabbit anti-Mas (1:100, NBP1-78,444, NOVUS), rat anti-iba1 (1:500, ab283346, Abcam), rabbit anti-NeuN (1:400, ab279297, Abcam), rabbit anti-GFAP (1:400, ab279291, Abcam), rabbit anti-MPO (1:100, ab208670, Abcam), rabbit anti-IL-1β (1:200, 12,426, Cell Signaling Technology), rabbit anti-CD16/32 (1:500, ab223200, Abcam), rabbit anti-iNOS (1:200, 18,985–1-AP, Proteintech), rabbit anti-Arg1 (1:200, 16,001–1-AP, Proteintech), rabbit anti-CD163 (1:200, 16,646–1-AP, Proteintech). Sections were then washed with PBS three times with 10 min intervals, and incubated with the corresponding secondary antibody for 1 h at room temperature. Nuclear staining was conducted by 4′,6-diamidino-2-phenylindole (DAPI). Sections were then visualized, and photographed under a Nikon microscope.
TUNEL staining
For quantification of neuronal apoptosis, double staining of neuron marker NeuN and TUNEL staining was conducted using One Step TUNEL Apoptosis Assay Kit (Beyotime) according to the manufacturer’s instructions at 3d after ICH. The number of TUNEL-positive neurons was counted manually in the peri-hematoma area. Data were expressed as the ratio of TUNEL-positive neurons (%).
FJC staining
At 3 days post ICH, degenerating neurons were assessed using a modified FJC Ready-to-Dilute Staining (Millipore) with FJC staining, as previously described [36]. According to the manufacturer’s instructions, the brain slices were baked in a 37 ℃ oven for 2–3 days, and consecutively incubated in solutions A, B, and C, then thoroughly washed. The slices were then baked at 50 ℃ for 10 min and incubated in xylene for 2 min. Once dried, the slices were sealed with neutral resin before being analyzed and photographed under a Nikon confocal microscope after a 5-min interval. The FJC-positive cells were counted in the peri-hematoma regions of the brain. The data were averaged and expressed as positive cells/mm2.
Nissl staining
In this study, the Nissl staining reagent (Servicebio) was utilized to evaluate neuronal degeneration in the hippocampus following ICH. Brain slices were washed in a container filled with PBS, and subsequently incubated with Nissl’s dye solution for 3 min. Then, the brain slices were rinsed with water until it ran clear and dried in an oven at 65 ℃. After being completely dried, the slices were carefully mounted with neutral resin and photographed with a microscope. Neuronal degeneration in the hippocampus cornu ammonis 1(CA1) region was observed and quantified.
Cell culture and vitro ICH model
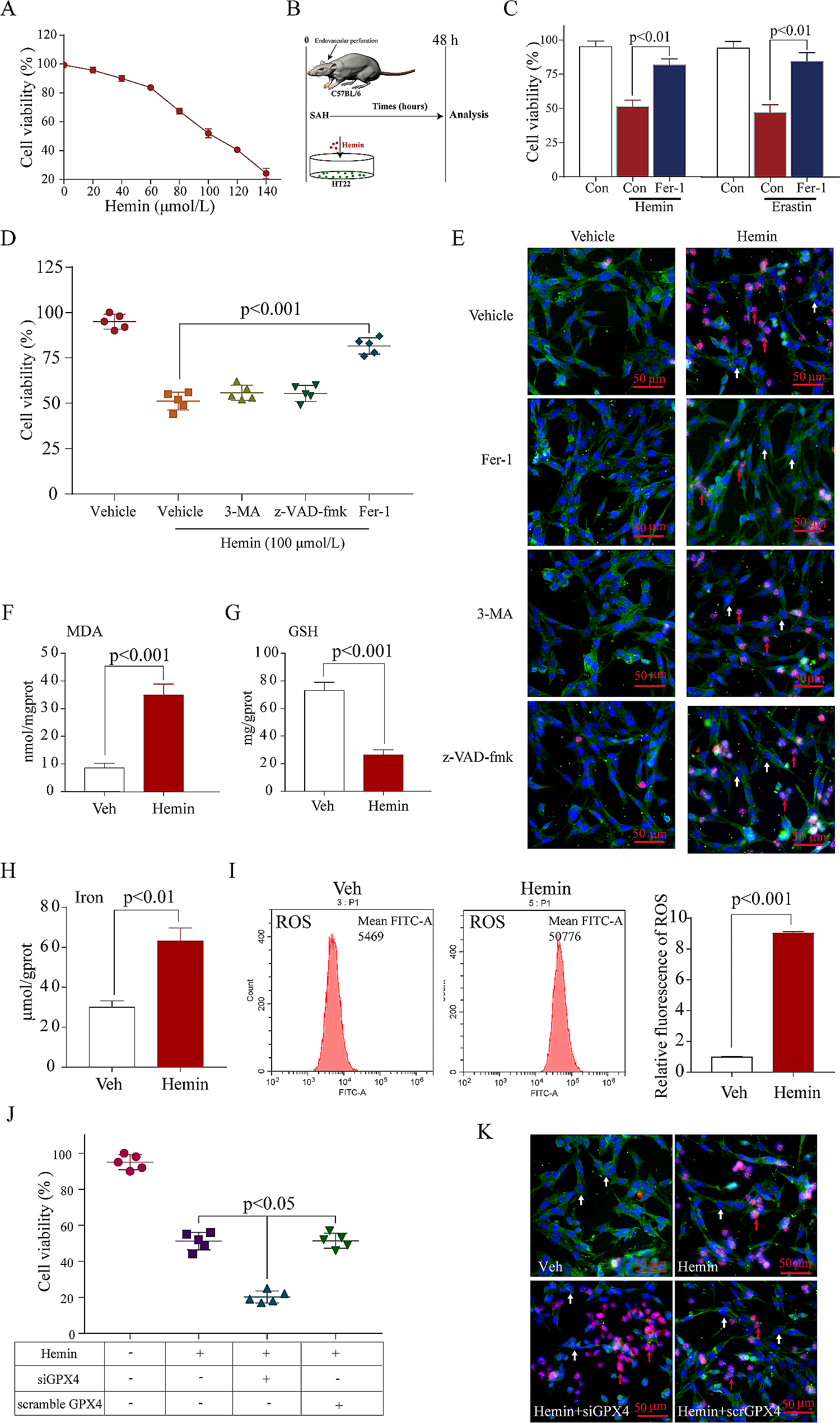
Microglial BV2 cells and neuronal HT22 cells were cultured in a complete medium containing DMEM with high glucose, 10% FBS and 1% penicillin/streptomycin at 37 °C under 5% CO2. Cells treated with a complete medium containing 10 μM hemin (Sigma-Aldrich, Germany) and vehicle for 24 h to mimic the ICH model in vitro. AVE0991-treated conditioned medium of BV2 cells were used to culture HT22 cells to elucidate whether AVE0991 influences neuronal apoptosis through microglial cells. Co-treatment with 10 μM AVE0991 was used to evaluate the protective effects of Mas activation.
Dead/live assay
The Calcein/PI Cell Viability/Cytotoxicity Assay Kit (Beyotime) was used to conduct dead/live experiment according to the instruction. HT22 cells were washed with PBS and then incubated with Calcein/PI working solution at 37 °C for 30 min darkness. Cells were washed again and observed under a fluorescence microscope.
Statistical analysis
All the data in this study were reported as the mean and standard deviation (SD). Results were analyzed by Student’s t-test, One-way ANOVA with Tukey’s post hoc test or Two-way ANOVA with Tukey's post hoc test where appropriate. A P-value < 0.05 was considered statistically significant.





留言 (0)