
記住我


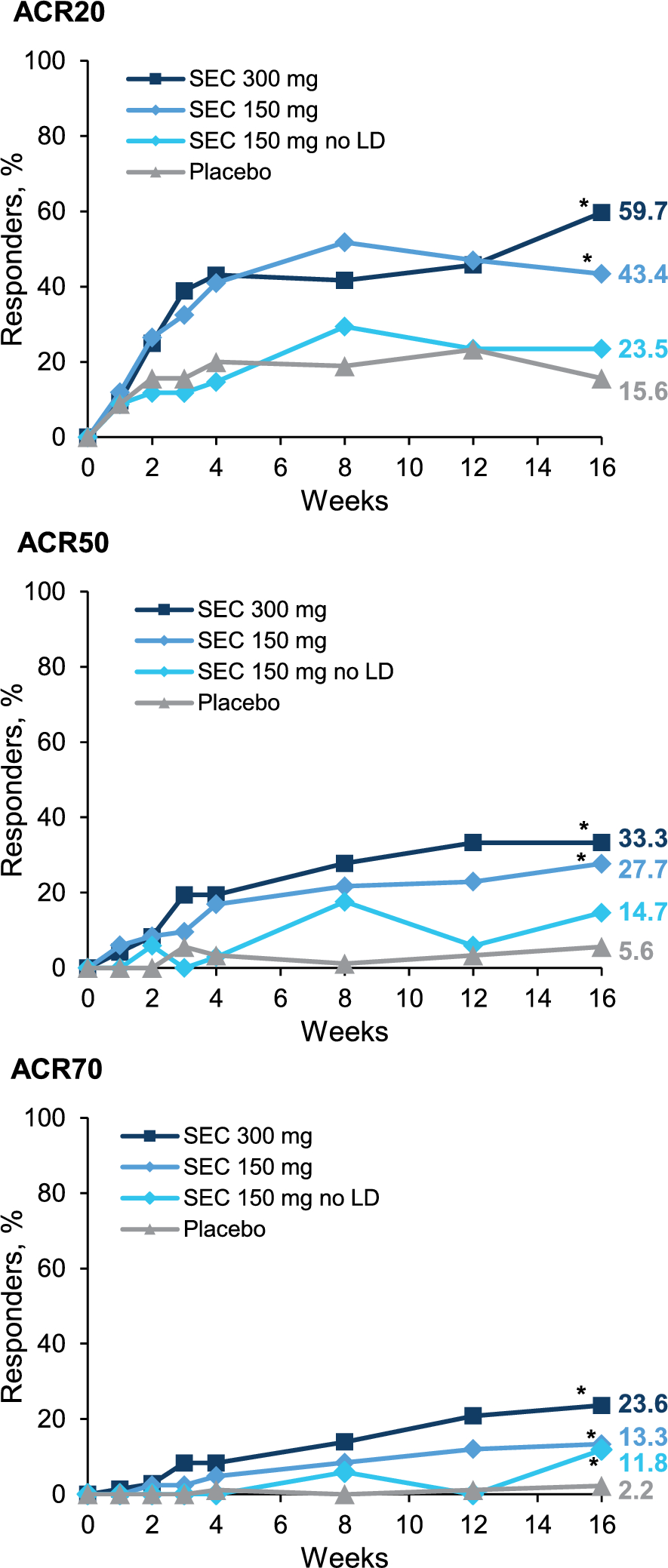
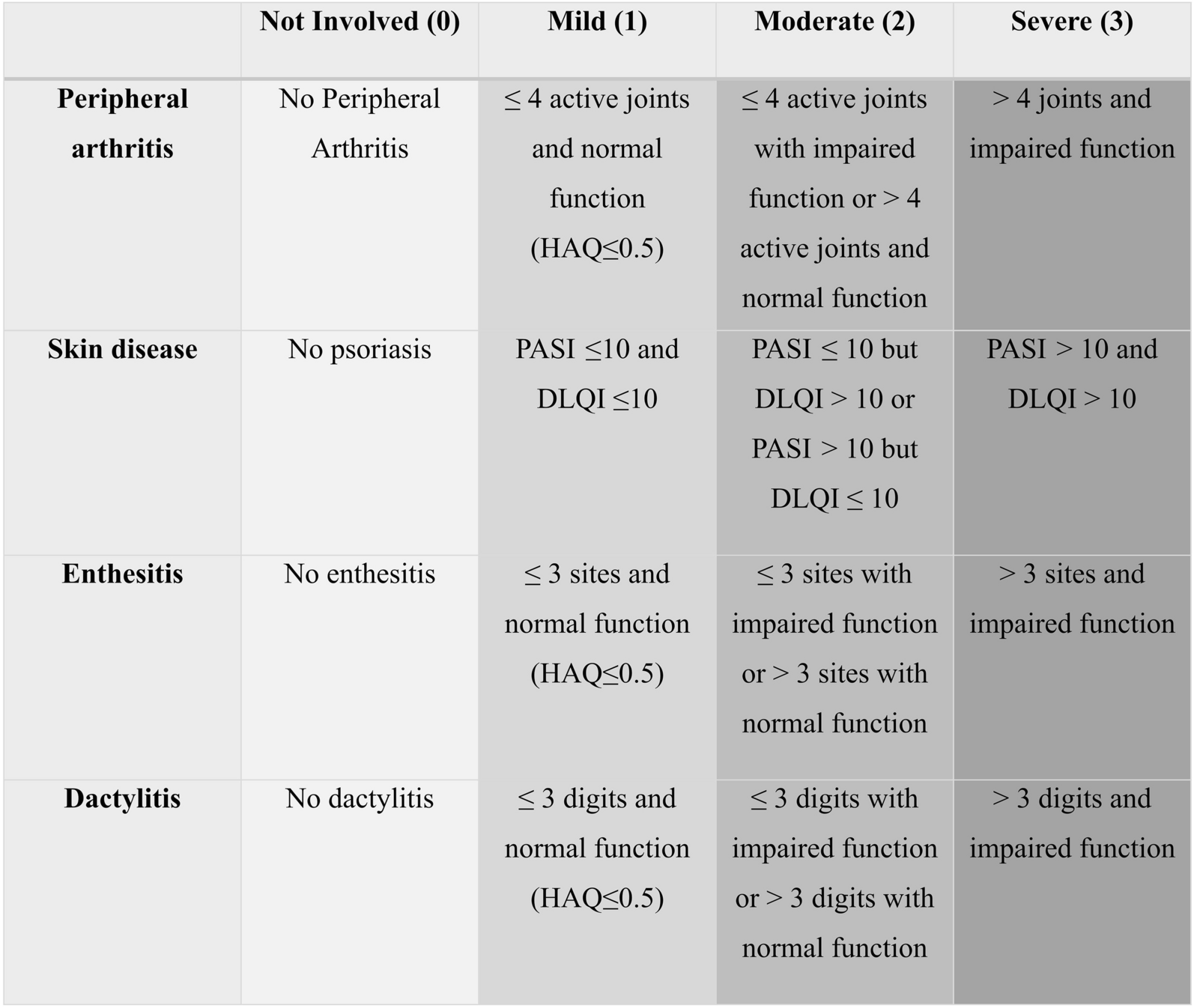
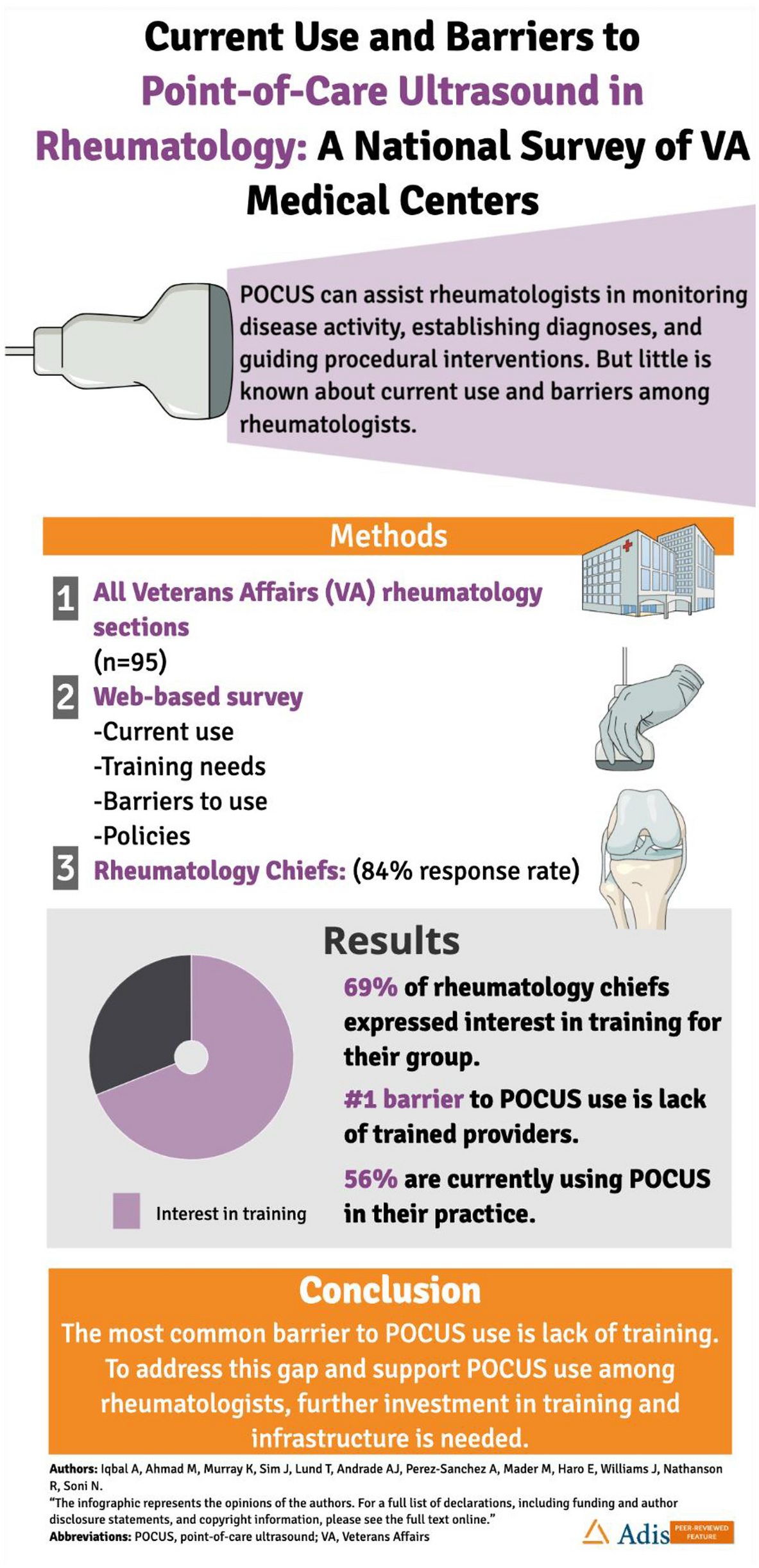
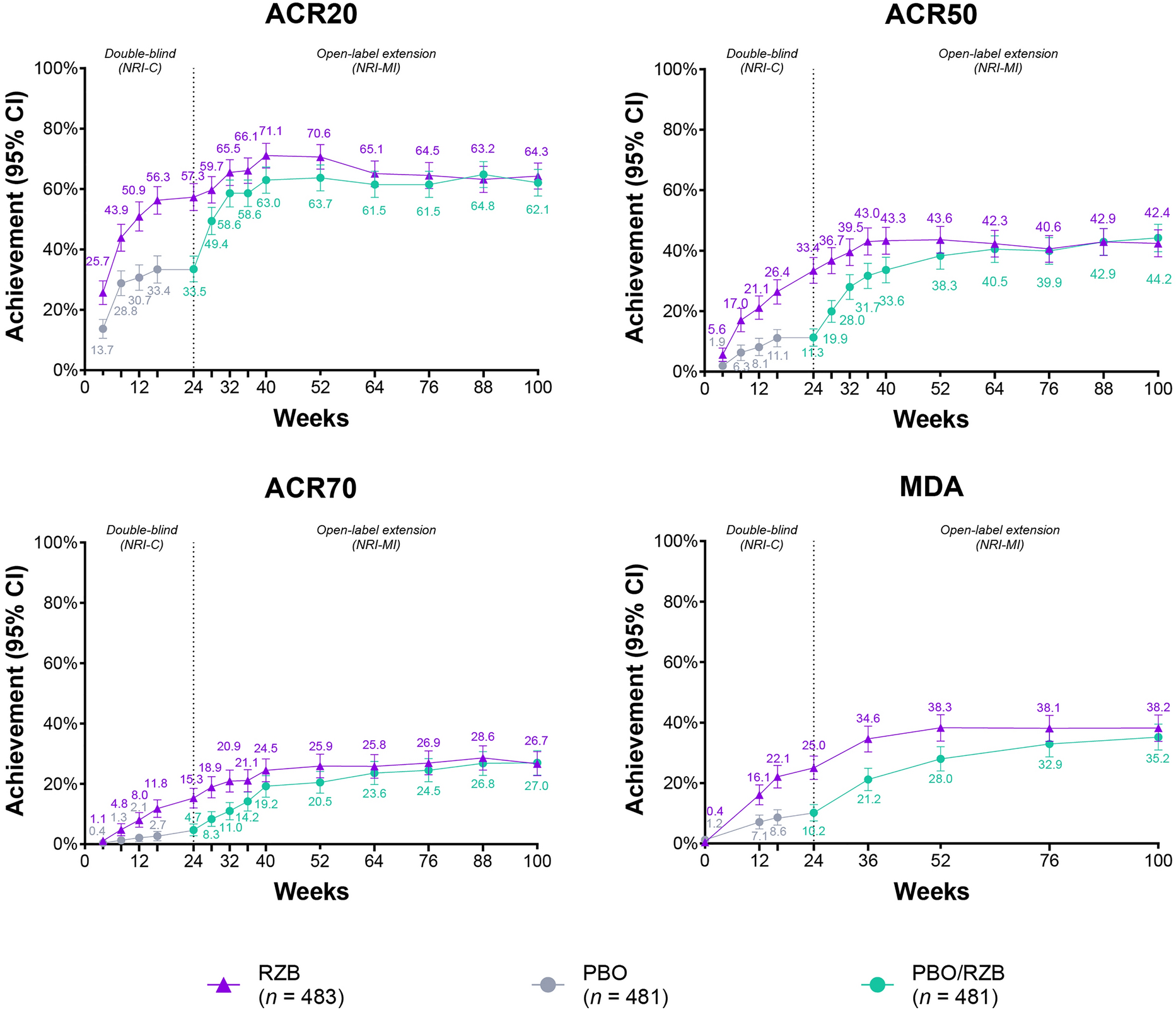
This phase 1a randomized, double-blind, placebo-controlled study (ISRCTN13175485) evaluated the safety, tolerability, and PK of a single IV or SC dose of RO7303509 in healthy participants. Participants were enrolled into six single-dose cohorts, consisting of eight participants each (6 active: 2 placebo), that tested the following doses of RO7303509: 50 mg, 150 mg, and 240 mg (IV) and 240 mg, 675 mg, and 1200 mg (SC) (Fig. 1).
Fig. 1
Study design. In each cohort, 2 additional participants were enrolled and treated with placebo (i.e., 6 participants, RO7303509; 2 participants, placebo). HV Healthy volunteers, SAD single ascending dose, IV intravenous, SC subcutaneous
Participants were screened within 35 days of randomization (day − 35 to day − 1) and checked into the clinical research unit (CRU) on day − 1. On day 1, participants were randomized and received a single dose of RO7303509 or placebo. For each cohort, two sentinel participants were dosed on the first day (1 active:1 placebo), and the remaining participants in each cohort were dosed at least 48 h after dosing of the sentinel pair, if no safety concerns arose. Participants remained in the CRU from day − 1 to day 3, and follow-up visits occurred through day 85. Dose escalation, including from IV to SC administration, proceeded if the safety information and emerging PK data through day 43 from the prior cohort were favorable.
The clinical study protocol and associated documents were approved by an Institutional Review Board before study initiation. The study was conducted in full conformance with the ICH E6 guideline for Good Clinical Practice and the principles of the Declaration of Helsinki. The study complied with the requirements of the ICH E2A guideline (Clinical Safety Data Management: Definitions and Standards for Expedited Reporting) and U.S. Food and Drug Administration (FDA) regulations and applicable local, state, and federal laws. All participants provided written, informed consent before enrolling in the study.
ParticipantsEligible participants were adults, 18–75 years old, with a body mass index (BMI) of 18–32 kg/m2, a body weight of 50–150 kg at screening, and healthy, as determined by medical history, physical examination, clinical laboratory tests, vital signs, and 12-lead electrocardiograms (ECG). Female participants were excluded if pregnant or breastfeeding. Potential participants were also excluded if they had treatment with an investigational therapy within 90 days before initiation of study drug; an infection requiring antibiotics within 14 days before screening or chronic infection requiring systemic treatment within 1 year before study drug initiation; a history of smoking or alcohol or drug abuse; were taking co-medications, except those approved by the medical monitor; a recent history (within 3 months) of root canal, unresolved dental infection, poor dentition, and/or dental implant placement; and any serious medical condition or abnormality that would preclude safe participation and completion of study.
Randomization and BlindingAfter eligibility requirements were confirmed, participants were randomized on day 1 via an interactive voice or web-based response system. Study site personnel and participants were blinded to treatment assignment during the study. Sponsor personnel were blinded to treatment assignment, except for individuals who required access to treatment assignments to fulfill their job roles during the trial. PK and immunogenicity samples were collected from placebo-treated participants to maintain blinding; laboratories responsible for PK and anti-drug antibody (ADA) analyses were unblinded to identify appropriate samples for analysis. Placebo doses were matched in appearance and administration to the respective IV or SC doses.
Safety Outcomes and AssessmentsThe primary outcome was the incidence and severity of AEs. AE terms were mapped to the Medical Dictionary for Regulatory Activities (MedDRA, version 25) thesaurus terms, and severity was determined according to the World Health Organization Toxicity Grading Scale. The study also monitored the change from baseline in targeted vital signs, targeted clinical laboratory test results, and 12-lead ECG parameters.
Pharmacokinetic Outcomes and AssessmentsThe secondary PK objective was to characterize the PK profile of a single dose of RO7303509 based on the serum concentration of RO7303509 at specified time points and the PK parameters.
Serum samples for PK analyses were collected on day 1 (predose and at 1, 2, 4, and 6 h after the start of infusion), at multiple timepoints throughout the study, and at the study completion visit on day 85. RO7303509 serum concentrations were measured using a sandwich enzyme-linked immunosorbent assay (ELISA), where capture antibody (clone 2A10.47; Genentech, Inc., South San Francisco, CA, USA) was coated on a microtiter plate to bind to RO7303509, followed by a biotinylated antibody (clone 2A10.37; Genentech Inc.) bound to the immobilized analyte and was detected by a horseradish peroxidase (HRP)-streptavidin conjugate.
Serum PK was summarized by estimating total exposure (area under the concentration–time curve; AUC), maximum serum concentration (Cmax), total clearance (CL) or apparent clearance (CL/F), volume of distribution (V) or apparent volume of distribution (V/F), time to maximum concentration (Tmax), terminal half-life (t1/2), and bioavailability (as appropriate for data collected). PK parameters were analyzed using non-compartmental methods in Phoenix WinNonlin (version 8.3, Certara, Princeton, NJ, USA).
Dose proportionality was assessed using a log-transformed power model. For full details see Eisenblaetter et al. [43].
Population Pharmacokinetic AnalysisA population pharmacokinetics (popPK) model was developed to describe serum RO7303509 concentration–time profiles. Various structural models were tested, including one-compartment, two-compartment, and three-compartment models, with the results showing that the PK of RO7303509 in the tested clinical dose range was best described by a two-compartment model. We used data from days 0–84 from the six cohorts in the study with single doses of RO7303509 (the IV cohorts received doses of 50, 150, 240 mg, and the SC cohorts received doses of 240, 675, 1200 mg). The PK population included participants who received at least one dose of RO7303509 and had at least one evaluable PK sample. The SC/IV popPK model included a depot compartment for SC with first-order SC absorption kinetics (Electronic Supplementary Material [ESM] Fig. 1). RO7303509 PK data were analyzed by nonlinear mixed-effects modeling (NONMEM) using the first-order conditional estimation method (FOCE). Models were evaluated based on the likelihood objective function value (OFV) provided by NONMEM (version 7.5.0; [44]), careful evaluation of goodness-of-fit plots, and precision of the parameter estimates (relative standard error [RSE]).
Immunogenicity Outcomes and AssessmentsThe secondary immunogenicity objective was to evaluate the immune response to RO7303509 by measuring the prevalence of ADAs at baseline and the incidence of ADAs during the study.
Serum samples for ADA analyses were collected on day 1, day 15, day 29, day 57, and at the study completion visit on day 85. ADA sample analysis was performed using a homogenous bridging ELISA. Biotin-conjugated drug and digoxigenin-conjugated drug were co-incubated overnight with diluted samples. The mixture was transferred to a streptavidin-coated high-binding plate, and the signal was detected using an HRP-conjugated murine anti-digoxin monoclonal antibody (Jackson ImmunoResearch Inc., West Grove, PA, USA).
When determining post-baseline incidence, subjects were considered to be ADA positive if they were ADA negative at baseline but developed an ADA response following study drug exposure (treatment-induced ADA response), or if they were ADA positive at baseline and the titer of one or more post-baseline samples was at least 0.60 titer units greater than the titer of the baseline sample (treatment-enhanced ADA response). Subjects were considered to be ADA negative if they were ADA negative or had missing data at baseline and all post-baseline samples were negative, or if they were ADA positive at baseline but did not have any post-baseline samples with a titer that was at least 0.60 titer units greater than the titer of the baseline sample (treatment unaffected).
Exploratory Outcomes and AssessmentsThe exploratory biomarker objective was to evaluate serum levels of the TGFβ pathway-associated biomarkers periostin and COMP after treatment with RO7303509 to provide evidence of RO7303509 activity. Samples for biomarker analyses were collected at baseline on day − 1, and after dosing on day 1, day 5, day 8, day 15, day 29, and day 85. Periostin was tested at Microcoat Biotechnologie GmbH (Bernried am Starnberger See, Germany) using the COBAS Elecsys system (Roche Diagnostics Corp., Indianapolis, IN, USA) with proprietary antibodies, and COMP was tested at Frontage Laboratories, Inc. (Exton, PA, USA) using the research-grade ProteinSimple Ella platform (SPCKB-PS-000756; ProteinSimple, San Jose, CA, USA). Absolute levels of periostin and COMP were graphed as the mean ± standard error (SE).
Dose SelectionWe selected the first-in-human dose based on a PK/pharmcodynamic (PD)-guided approach, leveraging minimal pharmacological activity of RO7303509 in a bleomycin-induced mouse model of pulmonary fibrosis. The activity of RO7303509 on TGFβ3-induced target gene expression determined the minimal pharmacologically active dose, and exposure from this dose was used to predict the first-in-human dose of 50 mg IV [42].
Drug AdministrationGenentech, Inc. provided the RO7303509 in solution. The solution used for IV administration was diluted in a manufacturer-provided diluent for injection and administered intravenously using syringe pumps. Undiluted study drug was administered by SC injection.
Ethical ApprovalThe clinical study protocol and associated documents were approved by an Institutional Review Board (Advarra IRB [Columbia, MD, USA] reference number: GA42285) before study initiation. The study was conducted in full conformance with the ICH E6 guideline for Good Clinical Practice and the principles of the Declaration of Helsinki. The study complied with the requirements of the ICH E2A guideline (Clinical Safety Data Management: Definitions and Standards for Expedited Reporting) and the U.S. FDA regulations and applicable local, state, and federal laws. All participants provided written, informed consent before enrolling in the study.
Statistical AnalysesAlthough no formal sample size calculations were performed, we screened a sufficient number of subjects to ensure eight participants at each dose level. Six participants dosed with active drug in each cohort was deemed sufficient to characterize safety, tolerability, and available PK data for RO7303509.
The safety analysis population included all participants who received one dose of study drug. The PK population comprised participants who received at least one dose of RO7303509 and had at least one evaluable PK sample. The immunogenicity analysis population included all participants with at least one post-dose ADA assessment. For all analysis populations, participants were grouped according to treatment received.
留言 (0)