
記住我


Overall, 964 patients were randomized, with 939 (97.4%) eligible patients choosing to enter period 2 to receive open-label risankizumab. Of all randomized patients, 828 (85.9%) were enrolled at the week 100 data cutoff date (continuous risankizumab, n = 412; placebo/risankizumab, n = 416). As previously reported, baseline characteristics were similar between treatment groups [11]. The most common reasons for study discontinuation in study period 2 were withdrawal of consent (n = 36, 3.7%) and < 20% improvement in tender/swollen joint count for two consecutive visits compared with baseline, as mandated by study protocol (n = 23, 2.4%).
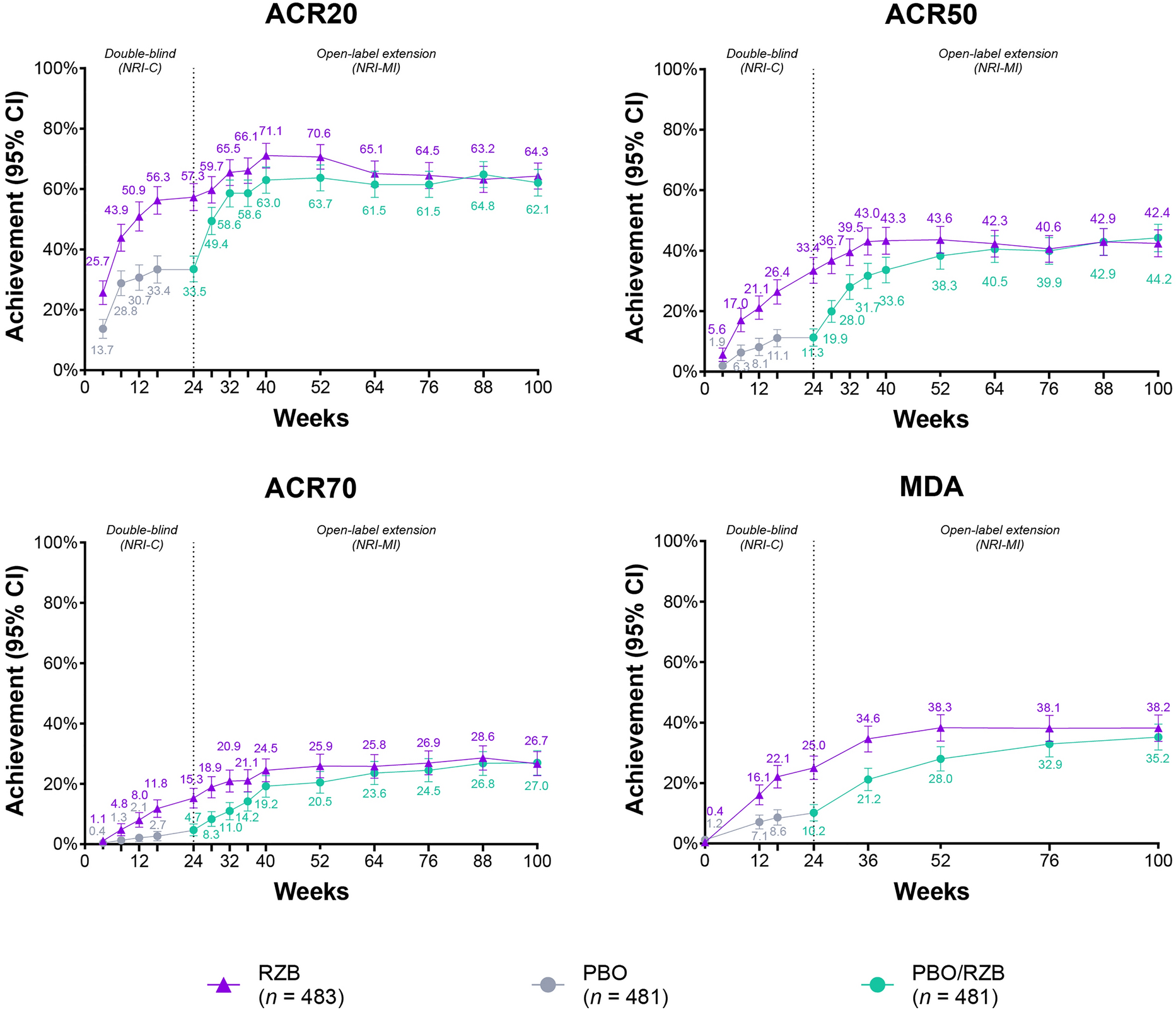
EfficacyJoint-Related Endpoints Through Week 100As previously reported, significantly more patients receiving risankizumab achieved the primary endpoint of ACR20 at week 24 (57.3%) compared with placebo (33.5%; p < 0.001; Fig. 1) [11].
Fig. 1
Achievement over time of ACR improvement endpoints and MDA. ACR20/50/70 ≥ 20%/≥ 50%/≥ 70% improvement in American College of Rheumatology criteria for symptoms of rheumatoid arthritis, CI confidence interval, MDA minimal disease activity, NRI-C, nonresponder imputation incorporating multiple imputations to handle missing data due to COVID-19, NRI-MI nonresponder imputation incorporating multiple imputations to handle missing data due to COVID-19 and geopolitical conflict, and all other missing data treated as nonresponders, including missing data and patients who received rescue therapy, PBO placebo, RZB risankizumab
At week 100, 64.3% and 62.1% of patients achieved ACR20 in the continuous risankizumab cohort and placebo/risankizumab cohort, respectively, which were consistent with results observed at week 52 in both groups (70.6% and 63.7%) (Fig. 1).
Durable efficacy was also achieved for ACR50 and ACR70 through week 100. The proportion of patients achieving ACR50 at week 100 was 42.4% in the continuous risankizumab cohort and 44.2% in the placebo/risankizumab cohort; the corresponding proportion of patients achieving ACR50 at week 52 was 43.6% and 38.3% and at week 24 it was 33.4% and 11.3%, respectively (Fig. 1) [11]. The proportion of patients achieving ACR70 was durable at week 100 (26.7% and 27.0%) from week 52 (25.9% and 20.5%); these proportions were greater than week 24 (15.3% and 4.7%) in both the continuous risankizumab and placebo/risankizumab cohorts (Fig. 1) [11].
Similar findings in ACR20, ACR50, and ACR70 were reported in both treatment cohorts when analyzed using the as observed data approach (Supplementary Material Fig. 1). In patients with ACR20 response at week 52, 81.2% of both cohorts maintained their response at week 100.
Maintenance of response at week 100 was also observed in 72.5% and 79.7% of week 52 ACR50 responders and in 69.1% and 71.2% of week 52 ACR70 responders in the continuous risankizumab and placebo/risankizumab cohorts, respectively (Supplementary Material Table 2).
Maintenance of response at week 100 for ACR20, ACR50, and ACR70 in week 52 responders was also seen in both cohorts when evaluated using the as observed approach (Supplementary Material Table 2).
Mean change from baseline in PsA-mTSS in the KEEPsAKE 1 study indicated low radiographic progression from baseline to week 100 (0.34 for the continuous risankizumab cohort and 0.45 for the placebo/risankizumab cohort), which remained stable from week 52 (0.21 and 0.27) and week 24 (0.23 and 0.32) in both treatment cohorts, respectively (Table 1). Most patients at week 100 (90.6% for continuous risankizumab and 86.9% for placebo/risankizumab) achieved change from baseline in PsA-mTSS ≤ 0, with no radiographic progression defined as PsA-mTSS ≤ 0 and PsA-mTSS ≤ 0.5, similar to week 52 (91.7% and 89.9%) and week 24 (92.4% and 87.7%) (Table 1). Similar numerical results were reported in the proportion of patients achieving change from baseline in PsA-mTSS ≤ 0.5 at week 100, with a comparable pattern of results across weeks 52 and 24 (Table 1). Similar findings in change from baseline in PsA-mTSS were reported when analyzed using as observed data approach (Supplementary Material Table 3).
Table 1 Efficacy assessmentsIn a prespecified pooled analysis of the KEEPsAKE 1 and KEEPsAKE 2 studies, most patients (57.7% for continuous risankizumab and 58.9% for placebo/risankizumab) with enthesitis at baseline achieved enthesitis resolution at week 100 (Fig. 2). Resolution of enthesitis was durable from earlier time points (week 52: 55.0% and 57.4%, respectively; week 24: 48.4% and 34.8%) (Fig. 2). Similarly, the majority of patients in the KEEPsAKE 1 and KEEPsAKE 2 studies with dactylitis at baseline experienced resolution at week 100 (continuous risankizumab, 76.2%; placebo/risankizumab, 75.3%), which was durable from week 52 (76.6% and 72.8%) and week 24 (68.1% and 51.0%) (Fig. 2).
Fig. 2
Achievement over time of resolution of enthesitis, resolution of dactylitis, improvement in PASI 90, and reduction in mNAPSI. PASI 90 reported only for patients with psoriasis-affected BSA ≥ 3 at baseline. mNAPSI reported only for patients with nail psoriasis at baseline. Resolution of enthesitis and dactylitis reported as pooled results from the KEEPsAKE 1 and 2 studies for patients with enthesitis (LEI > 0) or dactylitis (LDI > 0) at baseline. BSA body surface area, CI confidence interval, MMRM mixed-effect model for repeated measures, LDI Leeds Dactylitis Index, LEI Leeds Enthesitis Index, mNAPSI modified Nail Psoriasis Severity Index, NRI-C nonresponder imputation incorporating multiple imputations to handle missing data due to COVID-19, NRI-MI nonresponder imputation incorporating multiple imputations to handle missing data due to COVID-19 and geopolitical conflict, and all other missing data treated as nonresponders, including missing data and patients who received rescue therapy, PASI 90 ≥ 90% improvement from baseline in Psoriasis Area and Severity Index, PBO placebo, RZB risankizumab
Change from baseline in resolution of enthesitis and resolution of dactylitis was similar when analyzed using the as observed data (Supplementary Material Fig. 2).
Skin and Nail Clearance Endpoints Through Week 100In patients who had a psoriasis-affected BSA ≥ 3% at baseline, the proportion who achieved PASI 90 was improved in both the continuous risankizumab and placebo/risankizumab cohorts at week 100 (71.3% and 67.8%, respectively), and the proportion of patients was sustained from week 52 (67.9% and 60.7%) and increased from week 24 (52.3% and 9.9%) (Fig. 2) [11]. Findings were similar for change from baseline in PASI 90 for both cohorts when data were evaluated using an as observed data approach (Supplementary Material Fig. 2).
Reduction from baseline in mNAPSI scores by 14.3 points and 13.5 points was achieved at week 100 in the continuous risankizumab and placebo/risankizumab cohorts, respectively, which was durable from week 52 and increased compared with the week 24 results (Fig. 2) [11]. Change from baseline in mNAPSI results was similar for both cohorts when data were analyzed using an as observed data approach (Supplementary Material Fig. 2).
Reductions from baseline in PGA-F scores were observed with continuous risankizumab and with placebo/risankizumab cohorts at week 100 (− 1.4 and − 1.3, respectively), which were similar to the reductions observed at week 52 (− 1.2 and − 1.1) and increased compared with reductions at week 24 (− 0.8 and − 0.4) (Table 1). In addition, the proportion of patients who achieved “clear” or “minimal” nail psoriasis at week 100 (PGA-F score of 0 or 1) and improvement ≥ 2 grades was 69.9% in the continuous risankizumab cohort and 67.9% in the placebo/risankizumab cohort, which was more than the proportion of patients achieving “clear” or “minimal” nail psoriasis at week 52 (58.0% and 49.2%) and week 24 (37.8% and 15.9%).
MDA Achievement Through Week 100The proportion of patients achieving MDA at week 100 in the continuous risankizumab cohort was 38.2%, which was stable from week 52 (38.3%) and more than week 24 (25.0%) (Fig. 1) [11]. The proportion of patients in the placebo/risankizumab cohort achieving MDA was increased at week 100 (35.2%) compared with week 52 (28.0%) and week 24 (10.2%), supporting the durability of efficacy with risankizumab [11] (Fig. 1). The overall findings in achieving MDA were similar when data were evaluated as observed (Supplementary Material Fig. 1). Maintenance of MDA response at week 100 in week 52 MDA responders was achieved by 75.5% and 78.2% of the continuous risankizumab and placebo/risankizumab cohorts, respectively; maintenance of MDA response was similar when data were evaluated as observed (Supplementary Material Table 2).
Patient-Reported Outcomes and Health-Related Quality of Life Through Week 100Patients reported a reduction in HAQ-DI scores from baseline at week 100 in the continuous risankizumab cohort and the placebo/risankizumab cohort (− 0.41 and − 0.36, respectively), which was sustained from week 52 (− 0.41 and − 0.32) and greater than week 24 (− 0.31 and − 0.11) [11] (Fig. 3). At week 100, clinically meaningful reductions from baseline in HAQ-DI (≥ 0.35 decrease from baseline) were achieved by 54.9% of the continuous risankizumab cohort and 48.4% of the placebo/risankizumab population, which were similar to the proportions of patients at week 52 (57.9% and 46.4%) and increased from week 24 (50.3% and 27.9%) [11] (Table 1).
Fig. 3
Change from baseline over time in HAQ-DI. Reported using MMRM. CI confidence interval, HAQ-DI Health Assessment Questionnaire–Disability Index, MMRM mixed-effect model for repeated measures, PBO placebo, RZB risankizumab
Results for patient’s assessment of pain also showed evidence of durability, with 26.9- and 25.0-point reductions from baseline observed at week 100 for patients in the continuous risankizumab cohort and the placebo/risankizumab cohort, respectively, which were similar to week 52 (− 26.5 and − 22.6) and increased from week 24 (− 21.0 and − 10.2) [11] (Table 1). Maintenance of achievement of clinically meaningful improvements in patient’s assessment of pain was observed at week 100 in 80.5% and 81.0% of week 52 responders in the continuous risankizumab and placebo/risankizumab cohorts (Supplementary Material Table 2). Patients also reported improved health-related quality of life based on the SF-36 PCS; the continuous risankizumab cohort experienced an 8.4-point reduction from baseline, and the placebo/risankizumab cohort had a 7.5-point reduction from baseline at week 100, which was similar to the results at week 52 (8.4 and 7.3, respectively) and greater than the results at week 24 (6.5 and 3.2) (Table 1).
At week 100, FACIT-Fatigue scores improved from baseline by 7.8 for patients receiving continuous risankizumab and 6.9 for those receiving placebo/risankizumab, which were durable from week 52 (8.0 and 6.5, respectively) and greater than week 24 (6.5 and 3.9) (Table 1). When data were evaluated using an as observed approach, the results for mean changes from baseline in clinically meaningful reduction in HAQ-DI (Supplementary Material Fig. 3), patient’s assessment of pain VAS, SF-36 PCS, and FACIT-Fatigue were similar for both treatment cohorts (Supplementary Material Table 3).
SafetyLong-term safety data included 946 patients who received risankizumab (either during the double-blind or the open-label period regardless of their original randomization cohort), representing 1708.4 PY of exposure (Table 2). The long-term safety cutoff date included ≥ 100 weeks of exposure, with an exposure-adjusted event rate for any TEAEs in patients receiving risankizumab of 130.1 E/100 PY; these event rates decreased compared with week 24 (177.6 E/100 PY) (Table 2).
The most common TEAEs through week 100 (≥ 4.0 E/100 PY) were COVID-19 infections (8.0 E/100 PY; Table 2) and nasopharyngitis (4.3 E/100 PY). COVID-19-related TEAEs had increased from 0.4 E/100 PY at week 24, likely due to the global COVID-19 pandemic that occurred during the study. TEAEs leading to discontinuation of the study drug were 2.1 E/100 PY through week 100 (Table 2). The most common TEAE leading to discontinuation of the study drug through week 100 was psoriatic arthropathy with seven reported events (0.4 E/100 PY).
Serious TEAEs through week 100 occurred at a rate of 7.6 E/100 PY (Table 2). A total of 39 serious infections (2.3 E/100 PY) were reported, with COVID-19 or COVID-19 pneumonia accounting for 18 of these 39 events. Two major adverse cardiovascular events (0.1 E/100 PY) were reported between weeks 52 and 100 (Table 2), including one sudden cardiac death and one nonfatal myocardial infarction; both events were assessed as having no reasonable possibility of being associated with the study drug by the investigator. Through week 100, there were no cases of active tuberculosis, and no new opportunistic infections had occurred since the one event (< 0.1 E/100 PY) of opportunistic infection (oropharyngeal candidiasis) reported at the week 52 cutoff date [14] (Table 2). There were 43 events of hypersensitivity (2.5 E/100 PY; Table 2), with none of these events being classified as serious. A total of nine malignant tumors were reported (0.5 E/100 PYs), including two patients with nonmelanoma skin cancer (NMSC) and seven patients with malignant tumors excluding NMSC (Table 2). Between the week 52 data cutoff date and the week 100 data cutoff date, there were no new cases of NMSC, and three patients were reported with new malignant tumors excluding NMSC. As previously reported in an analysis of the week 52 data cutoff date, two deaths occurred, one patient with urosepsis on study day 96 [11] and one sudden death on study day 502 [14], both deemed unrelated to the study drug. The sudden death reported at the week 52 cutoff date occurred in a patient who died at home and had “natural causes” stated as the primary cause of death on the death certificate; this was later adjudicated as a sudden cardiac death and defined as a major adverse cardiovascular event in the week 100 data analysis. After that period and in the current analysis, four additional patients died, including two patients with COVID-19; one patient with complications related to acute leukemia; and one patient with anemia, diverticulosis, and multiorgan failure due to complications of a hemicolectomy. The deaths were deemed unrelated to the study drug.
留言 (0)