Strains and growth conditions
All strains used in this study were detailed in Table S1. E. coli DH5α strains were cultured at 37 °C and 200 rpm using LB complex medium (10 g/L tryptone, 5 g/L yeast extract, and 5 g/L NaCl) containing ampicillin (100 mg/L) when appropriate. S. cerevisiae was cultured in YPD complex medium (10 g/L yeast extract, 20 g/L peptone, and 20 g/L glucose) at 30 °C before transformation. After transformation, yeast cells were grown in appropriate synthetic complete (SC) medium minus the auxotrophic compound (FunGenome Company, Beijing, China) complemented by the plasmids. To prepare solid LB or YPD media, agar (20 g/L) was included.
Primers and plasmid construction
All plasmids used for the present study were detailed in Table S1, while all primers were presented in Table S2. All plasmid constructions were achieved with DNA assembler. To construct the gRNA delivery vector, the S. cerevisiae SNR52p promoter, the synthesized crRNA, 20-bp complementary region (N20) and SUP4t were assembled into the NotI/EcoRI site of pDB78 to generate pDB78-X (a generic term of all gRNA delivery vector and “X” represents the serial number). The approach was used to produce the pDB78-1 (pDB78-SNR52p-FAA1gRNA), pDB78-2 (pDB78-SNR52p-FAA4gRNA), pDB78-3 (pDB78-SNR52p-POX1gRNA), pDB78-4 (pDB78-SNR52p-SAM2gRNA), and pDB78-5 (pDB78-SNR52p-ADO1gRNA) vectors.
To construct the DmJHAMT methyltransferase-encoding vector, S. cerevisiae genomic DNA was used to amplify the TPI1p promoter, while the codon-optimized DmJHAMT gene fragments were synthesized by GenScript (Nanjing, China). The synthesized terminator named Tguo1, which is only 39 bp long and enables an ease of cloning using inexpensive oligos, was introduced into the primer P48 for amplifying the gene DmJHAMT (Curran et al. 2015). Therefore, the amplified DNA fragment had the terminator Tguo1 at the end.
The two TPI1p and DmJHAMT fragments were then assembled in the pESC-HIS vector using the BamHI/EcoRI site to produce the pESC-HIS-1 plasmid.
Genetic manipulation
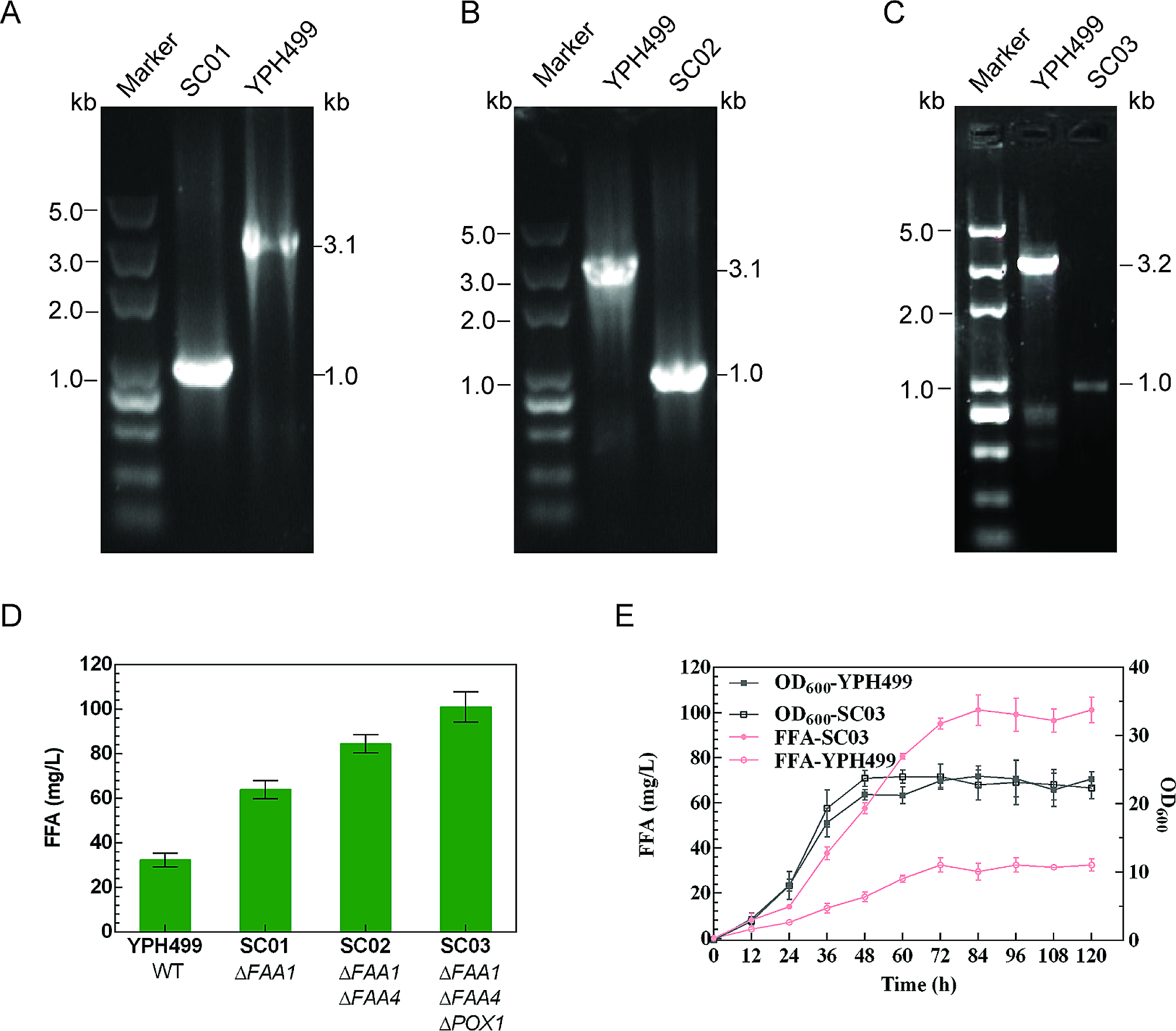
Homologous upstream and downstream arms (∼0.5 kb each) for target genes of interest were amplified from genomic DNA prepared from S. cerevisiae and jointed by overlap-extension PCR to be used as editing templates. Yeast transformation was performed using the lithium acetate method as described for S. cerevisiae (Gietz et al. 2002). A CRISPR/Cas9 approach was used for markerless genome editing. The schematic process of genome editing was shown in Fig. S1. Briefly, the Cas9 expression plasmid pCRCT was first introduced into the strain YPH499. After transformation, cells were plated on selective media (SC-uracil) and allowed to grow for 3–4 days until colonies appeared. Colony PCR was performed to verify the transformants. To obtain a knockout strain, the prepared gRNA delivery vector and editing template were introduced into Cas9 expressing cells. The resulting transformants were screened on selective media (SC-uracil and histidine). Eight to twelve colonies for each mutant were randomly selected and colony PCR was used to verify the success of genomic editing. Once the desired mutation was verified, the gRNA delivery vector containing the HIS1 marker was eliminated by culturing the strains in SC-uracil medium. The mutant could then be used for the next round of genome editing. When all gene manipulations were complete, the Cas9 expression vector containing the URA3 marker was eliminated by culturing the strains in YPD medium without selection pressure.
Flask fermentation
Individual colonies were used to inoculate 10 mL of SC or YPD media, followed by culture for ∼24 h at 30℃. After the preculture stage was complete, seed cultures were used to inoculate 100 mL of SC or YPD media in a 500 mL Erlenmeyer flask, adjusting the OD600 to 0.2 following inoculation. All assays were conducted in triplicate.
SAM assay
SAM extraction from fermentation broth was performed following the addition of 10% (w/v) perchloric acid with shaking a 220 rpm for 1 h at 30 °C. Lysates were then centrifuged (5 min, 13,000 rpm, 4 °C) to clarify the sample, after which high-performance liquid chromatography (HPLC) was used to analyze these samples with a C-18 column (Hypersil BDS 5 m 4.6 mm × 250 mm) (Chen and Tan 2018). These analyses were performed using a mobile phase consisting of 40 mmol/L NH4H2PO4, 2 mmol/L sodium heptyl sulfonate, and 18% methanol (Chen and Tan 2018). UV detection was performed at 254 nm. Triplicate analyses were conducted for all measurements. To measure the production of SAM, the standard curve for SAM was constructed. A series of standard solutions containing SAM (20, 40, 60, 120 and 240 mg/L) were analysed by HPLC. A least-squares fit was performed using the peak areas as the vertical coordinate and the concentration of SAM as the horizontal coordinate. SAM standard was purchased from Aladdin Ltd, Shanghai, China.
FFAs and FAMEs extraction
Total FFAs were extracted as described by Zhang et al. (Zhang et al. 2020). Briefly, 200 µL of cell culture was mixed with 10 µL 40% tetrabutylammonium hydroxide and 200 µL methylation reagent. The methylation reagent contained 200 mM methyl iodide as methyl donor. Prior to the methylation reaction, pentadecanoic acid was added as an internal standard to the mixture of methylation reagent and sample to a final concentration of 5 mg/L. The mixture was shaken for 45 min and then centrifuged at 6000 g for 5 min. 100 µL of the dichloromethane layer was then transferred to glass GC vials for subsequent analysis. FAMEs were extracted as described by Sherkhanov et al. (Sherkhanov et al. 2016). Briefly, 5 mL of cell culture was mixed with 6 mL of a 2:1 chloroform/methanol mixture.
FFAs and FAMEs quantification
A GC-FID approach was used to quantify levels of FFAs and FAMEs with an HP 5890 Series II gas chromatograph equipped with an HP-Innowax Column (0.32 mm × 30 m × 0.25 μm, Agilent). The injection volume for all samples was 1 µL, and other analytical parameters were as follows: inlet temperature 250 °C, split ratio 1:1, helium carrier gas at a 5 mL/min flow rate. The oven temperature was initially set to 160 °C for 3 min, after which it was increased at 5 °C/min to 255 °C and held for 3 min. The inlet temperature was 270 °C, while the detector temperature was 330 °C (Sherkhanov et al. 2016). FAME products were identified via a GC/mass spectrometry approach with an Agilent 6890 − 5975 instrument equipped with HP-Innowax Column (0.32 mm × 30 m × 0.25 μm, Agilent). Peaks were identified by comparing the results with GC retention times, known standards, and mass spectra included in the National Institute of Standards and Technology (NIST) database.
To measure the production of fatty acids and FAMEs, standard curves for the five kinds of FAMEs (C14:0, C16:0, C16:1, C18:0 and C18:1) were constructed. Briefly, a series of standard solutions containing five mixed FAME standards and internal standard methyl pentadecanoate (5 mg/L) were analysed by GC. In the standard solutions, the concentration gradients of each FAME were 1, 5, 10, 15, 20, 25 mg/L. A least-squares fit was performed using the ratio of the peak areas of the FAMEs to the peak areas of the internal standard as the vertical coordinate and the concentration of the FAME standards as the horizontal coordinate. FFA and FAME levels were analyzed by comparing the results to standard curves. FAME standards were purchased from Sigma-Aldrich.


留言 (0)