Collection of tissue samples
The study conformed to the STROBE guidelines. Participants must be over 18 and in good health. Patients were excluded who had blood diseases, a history of Hepatitis B or C, HIV, active malignancy, recent treatment with certain medications, or autoimmune disorders within 1 year. Oral health evaluation was conducted alongside systemic screening to ensure that only individuals without soft tissue lesions and no indication of oral or dental infection were included in the study. The inclusion criteria for OSF were clinical stage III to IV (< 20 mm mouth opening and/ or accompanied by leukoplakia or OSCC). The tissue samples used for analysis in this study were obtained from 19 participants who were diagnosed with OSF in the last three years. In addition, samples of standard human oral mucosa were collected from six patients who were either receiving treatment for mucosal hyperplasia or undergoing tissue resection as part of orthognathic surgery procedures. One fresh tissue sample from each group was sent for scRNA-seq analysis, while the remaining samples were made into sections, stained, and observed. T cells were sourced from four additional blood samples. It is important to note that all tissue samples were histopathologically diagnosed by the Pathology Department of the Air Force Medical University (AFMU) in Xi’an, Shaanxi Province, China. Detailed, written, informed consent was obtained from all volunteers in accordance with protocols approved by the ethical committee of the AFMU (IRB-REV-2022049; IRB-REV-2022058). Supplementary Table 1 provides information on the clinical parameters of the healthy volunteers and patients with OSF.
Preparation of samples for scRNA-seq
Upon collection, tissue samples were transported in sterilized culture dishes containing 10 mL of 1× Dulbecco’s Phosphate-Buffered Saline (DPBS; Thermo Fisher Scientific, Cat. no. 14190144) under chilled conditions to wash away residual tissue storage solution. The tissues were subsequently minced under similar cold conditions. A combination of 0.25% Trypsin (Thermo Fisher Scientific, Cat. no. 25200-072) and 10 µg/mL of DNase I (Sigma, Cat. no. 11284932001) were employed, each dissolved in phosphate-buffered saline (PBS) containing 5% Fetal Bovine Serum (FBS; Thermo Fisher Scientific, Cat. no. A3161001C), to facilitate tissue digestion. Human tissues underwent dissociation at a controlled temperature of 37 ℃, with an oscillation speed of 50 rpm maintained over approximately 40 min. To optimize cell yield and viability, the dissociated cells were intermittently harvested every 20 min. The resultant cell suspensions were filtered through a 40 μm nylon cell strainer. Furthermore, any red blood cells present were eliminated using 1× Red Blood Cell Lysis Solution (Thermo Fisher Scientific, Cat. no. 00-4333-57). Dissociated cells were rinsed with 1× DPBS incorporating 2% FBS. To assess cell viability, cells were stained with 0.4% Trypan blue (Thermo Fisher Scientific, Cat. no.15250061) and enumerated using a Countess® II Automated Cell Counter (Thermo Fisher Scientific, Waltham, MA, USA).
Preparation of the 10× library and the sequencing procedure
Each bead was characterized by a unique molecular identifier (UMI) and cell barcodes were loaded close to the saturation point. This ensured that each individual cell was suitably paired with a bead in a Gel Beads-in-Emulsion setup. Following exposure to a cell lysis buffer, polyadenylated RNA molecules were hybridized with the beads. The beads were subsequently collected into a single tube for subsequent reverse transcription. During cDNA synthesis, each cDNA molecule received a tag on its 5' end, which corresponds to the 3' end of a messenger RNA transcript. This tagging mechanism incorporated a UMI and cell label to trace the cell of origin for each molecule. The 10× beads were then subjected to second-strand cDNA synthesis, adapter ligation, and universal amplification. Sequencing libraries were assembled using the products of randomly interrupted whole-transcriptome amplification. This approach facilitated the enrichment of the 3' end of the transcripts, which were linked with the cell barcode and UMI. Following these stages, all remaining procedures, including library construction, were performed according to the standard manufacturer’s protocol (10× Genomics, Pleasanton, CA, USA). Sequencing libraries were quantified using a High Sensitivity DNA Chip (Agilent, Santa Clara, CA, USA) on a Bioanalyzer 2100 instrument with the Qubit High Sensitivity DNA Assay (Thermo Fisher Scientific). Finally, the prepared libraries were sequenced on a NovaSeq6000 (Illumina, San Diego, CA, USA) platform, utilizing 2 × 150 chemistry.
Processing and quality control of the scRNA-seq data
Reads were handled using the Cell Ranger 2.1.0 pipeline (10× Genomics), applying default and suggested parameters. FASTQ files generated from Illumina sequencing output were analyzed using the STAR algorithm [17]. Gene-barcode matrices were generated for each distinct sample through UMI counting and the filtering of non-cell associated barcodes. Consequently, a gene-barcode matrix was constructed, encompassing barcoded cells and corresponding gene expression counts. This output was then incorporated into the Seurat (v2.3.0) R toolkit to enable quality control and further analysis of the scRNA-seq data [18]. All functions were executed using default parameters, except where explicitly stated otherwise. Cells exhibiting fewer than 200 or more than 6000 detected genes (each gene necessitating at least one UMI aligned in a minimum of three cells) were excluded. The expression of mitochondrial genes was computed utilizing the PercentageFeatureSet function of the Seurat package [18]. Cells with mitochondrial gene expression exceeding 10% were excluded to eliminate low-activity cells. Data normalization (via the NormalizeData function in the Seurat package) was performed to extract a subset of variable genes, which were then identified while controlling for the strong correlation between variability and average expression. Data from diverse samples were subsequently integrated, following the identification of ‘anchors’ between datasets utilizing FindIntegrationAnchors and IntegrateData in the Seurat package [18]. Principal Component Analysis (PCA) was performed, and the data were reduced to the top 30 PCA components following scaling. The resulting clusters were visualized on a two dimensional (2D) map constructed using t-distributed Stochastic Neighbor Embedding (t-SNE).
Identification of cell types and subtypes via nonlinear dimensional reduction (t-SNE)
Cells were clustered using graph-based clustering of the PCA-reduced data, following the computation of a shared nearest neighbor graph using the Louvain Method [18]. For sub-clustering, the same procedure (scaling, dimensional reduction, and clustering) was applied to a specific dataset, typically restricted to one cell type. For each cluster, the Wilcoxon Rank-Sum Test was employed to identify significantly differentially expressed genes when compared with the remaining clusters. Cell types were then ascertained using SingleR and known marker genes [19].
Pseudotime analysis
Pseudotime analysis was conducted utilizing the Monocle2.14.0 software [20]. This analysis incorporated all subsets of epithelial cells and facilitated cell order sorting using the subgroup markers procured from previous analyses. This allowed for a holistic evaluation of the expression change relationship between subsets. The DDRTree algorithm was employed to reduce the dimension of known differentiation-related genes, genes exhibiting significant coefficients of variation, and the differential gene data of subsets. The differentiation time for each cell was calculated, a starting point was randomly selected, and the cells were sorted accordingly. Differentiation-related genes were then calculated based on the time of differentiation.
Cell–cell interaction analysis
To visualize and analyze intercellular communications derived from scRNA-seq data, a cellchat analysis was carried out [21]. A new cellchat object was created from the existing Seurat object, with cell types added to the cellchat object as cell metadata. Cellchat identified differentially overexpressed ligands and receptors for each cell group and linked each interaction with a probability value, thereby quantifying communications between two cell groups mediated by these signaling molecules. Significant interactions were recognized based on a statistical test that randomly permuted the group labels of cells and recalculated the interaction probability. A ligand or receptor was classified as ‘expressed’ in a particular cell cluster if it was present in more than 25% of cells.
Immunohistochemistry and immunofluorescence staining of tissue
For immunohistochemistry, tissue sections were deparaffinized, rehydrated, and antigen retrieval was performed using a citrate buffer. Sections were blocked with a protein-blocking solution and incubated with primary antibodies overnight at 4 °C and then with horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature. The 3,3′-Diaminobenzidine (DAB) substrate was added until the desired stain intensity developed. The antibodies used in this study recognized keratin 19 (KRT19) (1:1000, 60187-1-lg, Proteintech), alpha smooth muscle actin (α-SMA) (1:300, GTX100034, GeneTex), CD31 (also known as platelet and endothelial cell adhesion molecule 1 (PECAM1)) (1:200, GB113151, Servicebio), and CD3 (1:1000, 17617-1-AP, Proteintech). For immunofluorescence staining, tissue sections were deparaffinized, rehydrated, blocked, and then incubated with primary antibodies overnight at 4 °C. Fluorescently tagged secondary antibodies were then applied. To visualize cell nuclei, sections were counterstained using 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen). Digital images were captured utilizing a fluorescence microscope (Nikon A1R; Nikon Corporation, Tokyo, Japan). The integrated fluorescence intensity across these images was quantified using Image Pro Plus 6.0 software (Media Cybernetics, Rockville, MA, USA). The antibodies employed in this study included those recognizing KRT19 (1:500, 60187-1-lg, Proteintech), macrophage migration inhibitory factor (MIF) (1:300, 20415-1-AP, Proteintech), CD3 (1:300, 17617-1-AP, Proteintech), CD74 (1:500, GTX110477, Proteintech), and C-X-C motif chemokine receptor 4 (1:100, 60042-1-Ig, Proteintech).
Isolation and activation of T cells
In this study, peripheral blood mononuclear cells (PBMCs) were retrieved from the venous blood of healthy volunteers using Ficoll density gradient centrifugation. To isolate T cells, a specific number of PBMCs were resuspended in 100 µL of separation buffer and subjected to sorting using the MojoSort Human CD3 T Cell Isolation Kit (BioLegend; Cat. no. 480022). To sort the CD3 + T cells, 10 µL of CD3 negative antibody was added to each 1 × 107 cells. Once sorted, the cells were transferred to X-VIVO15 medium (serum-free hematopoietic cell medium) supplemented with 5% human serum for T cell culture. To activate the CD3 + T cells, CD3/CD28 magnetic beads (Dynabeads Human T-activator CD3/CD28, Life Technologies, Cat. no. 11131D) were employed. The activated CD3 + T cells were then utilized in experiments conducted a week later.
Cell culture
Various types of cells were used in the experimental procedures, including human oral epithelial keratinocytes (HOK-16B, zl-040906, ZLZT, Wuhan, China) and human gingival fibroblasts (HGFs, CP-H205, Procell). T cells and human DPSCs were also employed. DPSCs were sourced from Beijing SH Biotechnology (Beijing, China; http://www.bjshbio.com/) and their isolation and culture followed established methods [22, 23]. Cells (HGFs and DPSCs) from passages 4–6 were used to ensure consistency and reliability. Additionally, all cell lines were confirmed to be free of mycoplasma infection. To create the Epi1.2 cell model, HOK cells were cultured in an environment with 60 µg/mL arecoline for 24 h. Furthermore, all cell cultures were maintained in a 5% CO2 atmosphere at 37 °C to support optimal growth and functionality.
Cell migration assay
HOK cells were propagated to confluence in a 6-well plate, after which a sterile 10 µL pipette tip was used to introduce a scratch down the center of each well. Detached cells were cleared via washing, and the cells were subsequently incubated in fresh medium. Images were captured at the 0, 24, and 48 h time points using an inverted microscope, which permitted an evaluation of cell migration into the scratched area. The formula used was [Wound healing percentage = (initial wound area−wound area at a certain point in time) / initial wound area] [24].
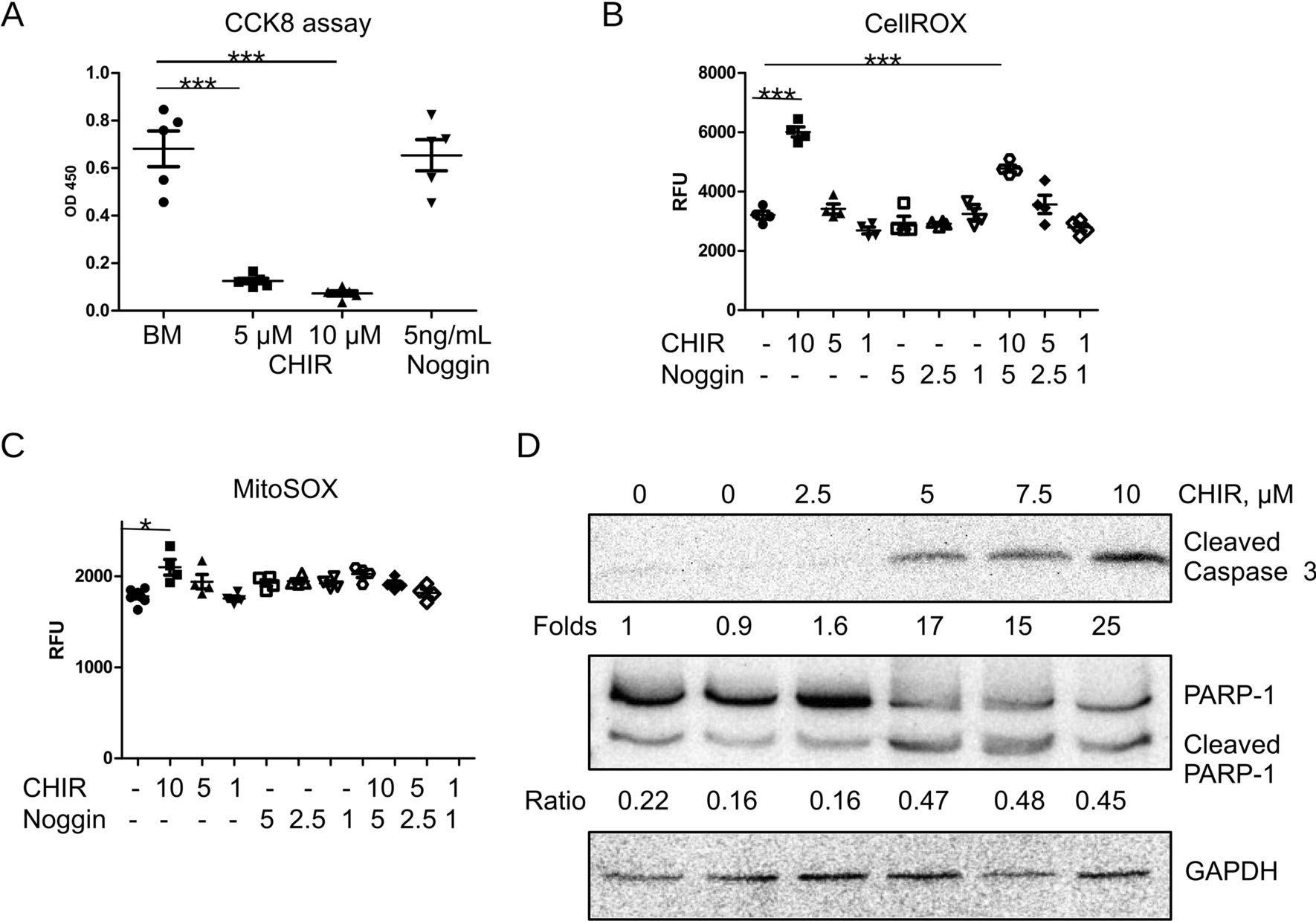
Cell proliferation assay
Human oral epithelial cells were sown at a density of 2,000 cells per well in a 96-well plate and incubated for 24 h and 48 h. Following this, the culture medium was replaced with a medium reflecting different conditions (Control: Dulbecco’s modified Eagle’s medium (DMEM); Arecoline: medium containing varying concentrations of arecoline (20, 40, 60, and 80 µg/mL), respectively). The cell proliferation rate was evaluated at the 0, 24, and 48 h, using a Cell Counting Kit-8 assay (Elabscience; Cat. no. E-CK-A362). The absorbance at 450 nm was recorded using a microplate reader (BIO-TEK, Winooski, VT, USA).
Immunofluorescence assay
Cells were fixed with 4% paraformaldehyde for 30 min. Following this, the cells were incubated with serum for 30 min to block non-specific binding sites. The primary antibody was added and incubated overnight after removal of the serum. The secondary antibody was then introduced and allowed to interact with the cells for 1 h, shielded from light. DAPI was subsequently added to stain the cell nuclei. The specimens were then observed using confocal scanning laser microscopy (Nikon A1R; Nikon Corporation). Integrated fluorescence intensity was quantified using ImageJ software (National Institute of Health, Bethesda, MD, USA). The antibodies employed in this study recognized KRT19 (1:300, 60187-1-lg, Proteintech), MIF (1:300, 20415-1-AP, Proteintech), CD3 (1:100, 17617-1-AP, Proteintech), CD74 (1:500, GTX110477, Proteintech), C-X-C motif chemokine receptor 4 (1:100, 60042-1-Ig, Proteintech) and α-SMA (1:500, GTX100034, GeneTex).
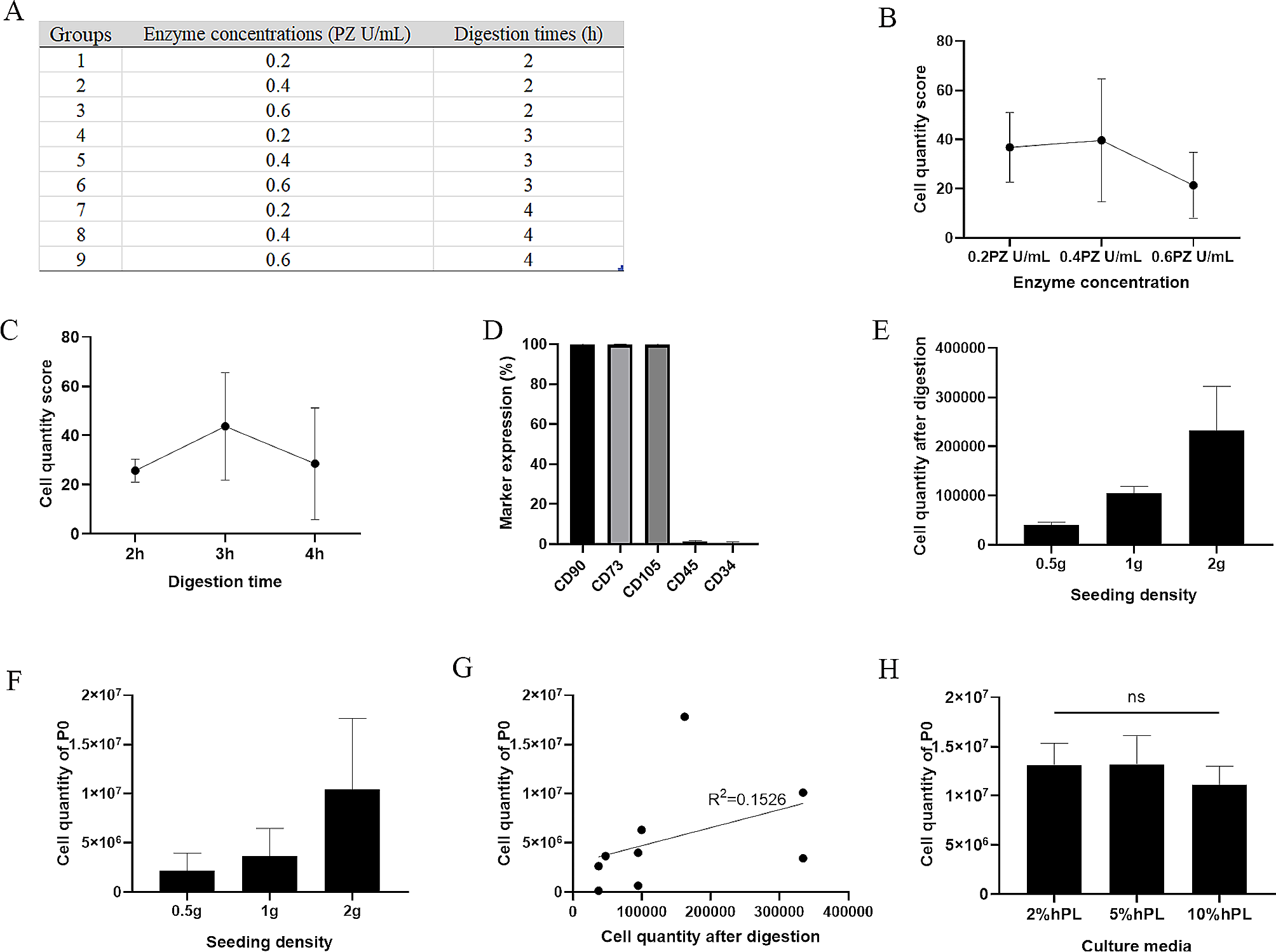
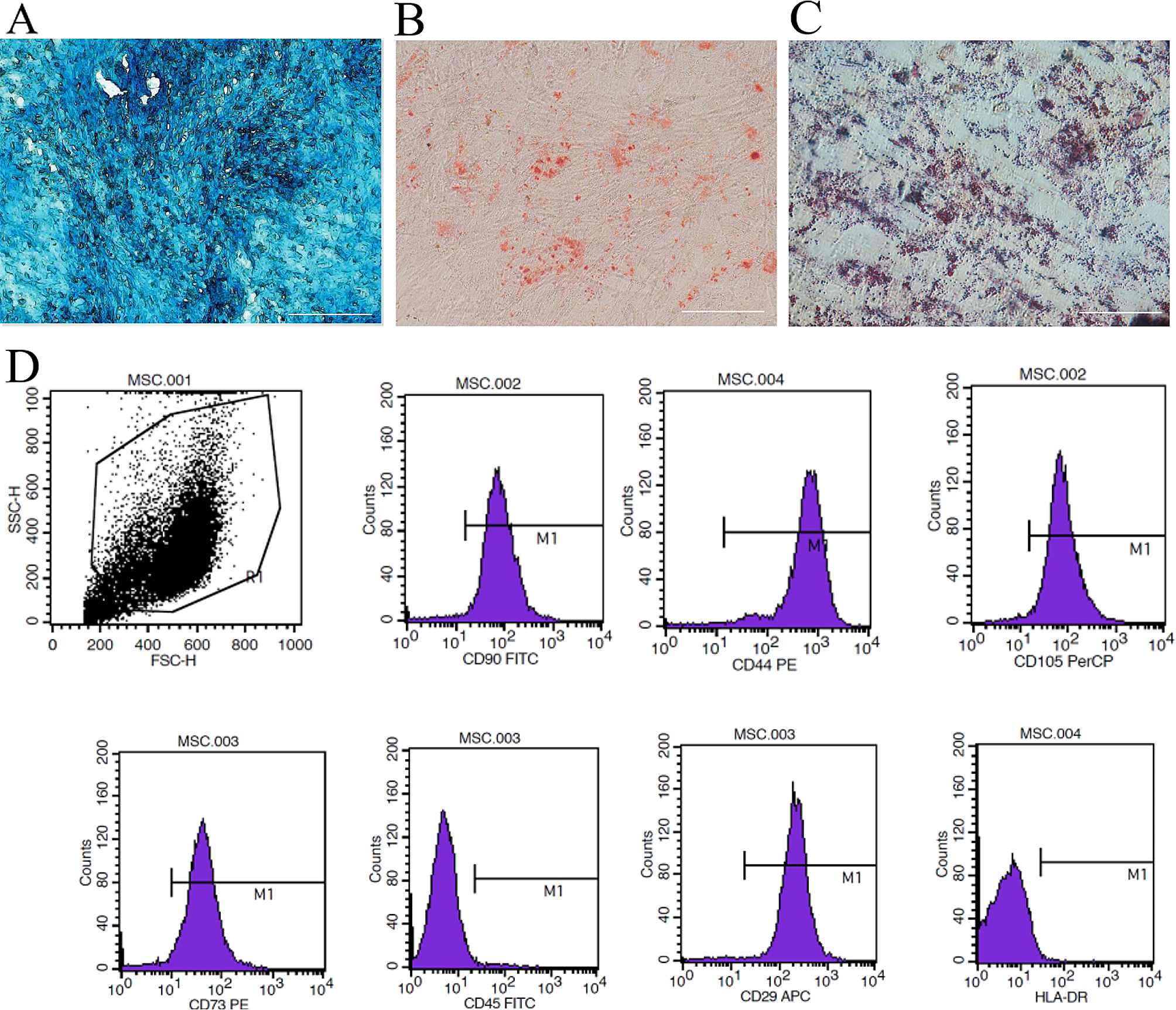
Flow cytometry assay
Human T cells, T cells co-cultured with Epi1.2 cells, and DPSCs were digested into single cell suspensions. T cell activation was assessed by flow cytometry staining for cell-surface expression of CD69 and CD25. Intracellular flow cytometry staining of granzyme B was conducted by fixing and permeabilizing cells with Cytofix/Cytoperm Fixation/Permeabilization Solution Kit (BD Biosciences; Cat. no. 554714). To determine the number of proliferating T cells, T cells and T cells co-cultured with Epi1.2 cells were incubated for 30 min at 37 °C in the dark with CytoTell Red 650 (AAT Bioquest, Cat. no. 22255). The following antibodies were used for flow cytometry staining: phycoerythrin (PE)-anti-human CD25 (Biolegend, 302606), Allophycocyanin (APC)-anti-human CD69 (Biolegend, 310910), fluorescein isothiocyanate (FITC) anti-human/mouse Granzyme B (Biolegend, 396404). For DPSC phenotype identification, anti-human monoclonal antibodies targeting specific surface markers were used, including PE-anti-human CD73 (BioLegend, 344,003), PE-anti-human CD90 (BioLegend, 328109), PE-anti-human CD105 (BioLegend, 323205), PE-anti-human CD11b (BioLegend, 982606), PE-anti-human CD34 (BioLegend, 343505), PE-anti-human CD45 (BioLegend, 304007), FITC-anti-human CD19 (BioLegend, 302205), and FITC-anti-human CD3 (BioLegend, 300,306). Following incubation, the cells were resuspended and kept in the dark. Flow cytometry (Coulter-XL; Beckman Coulter, Indianapolis, IN, USA) was used to detect the cell surface markers, and the data analysis was performed using FlowJo 10.0 software (Flow Jo LLC, Ashland, OR, USA) or EXPO32 ADC Analysis software (Beckman Coulter).
Transwell assay
These experiments were performed in 12-well Transwell plates with a 0.4 μm pore membranes (NEST; Cat. no. 724101). Human CD3 + T cells (1 × 106 ) were seeded to the upper compartment of the chamber, while Epi1.2 cells (2 × 106) were seeded to the lower compartment. Cells were cultured for 24 h and the number of T cells in the lower chamber was recorded for analysis.
In vitrosuppression assays
HOK cells were seeded in 6-well plates at 2 × 106 /mL. After cell adherence, 60 µg/mL arecoline medium was added to three wells and cultured for 24 h to induce Epi1.2 cells. The MIF inhibitor ISO-1 (1 µM) (HY-16692, Med Chem Express) was dissolved in dimethyl sulfoxide according to the manufacturer’s instructions and added into the cell culture medium. After incubation for 24 h, T cells (1 × 106) were added to the 6-well plates. Immunofluorescence and flow cytometry were used to detect Epi1.2 cells and T cells after 24 h of co-culture.
Enzyme-linked immunosorbent assay (ELISA)
In this study, HOK cells, Epi1.2 cells, T cells, and Epi1.2 + T cells were cultivated in 48-well plates for 24 h. Subsequently, the supernatant from each group was collected and used to detect various cytokines, including interleukin (IL)-6 (Servicebio, GEH0001), IL-1β (Servicebio, GEH0002), IL-17 (MultiSciences; EK117/2–96), transforming growth factor beta (TGF-β) (MultiSciences, EK981-96), tumor necrosis factor alpha (TNF-α) (Servicebio, GEH0004), and interferon gamma (IFN-γ) (Servicebio, GEH0006). The quantification of these cytokines was performed using ELISA kits according to the manufacturer’s guidelines. The absorbance at 450 nm was measured using the microplate reader.
Quantitative real-time reverse transcription polymerase chain reaction (qRT-PCR)
The supernatants of HOK cells, Epi1.2 cells, T cells, Epi1.2 + T cells and ISO-1 + Epi1.2 + T cells were collected, subjected to high-speed centrifugation, and the supernatants were co-cultured with HGFs. Total RNA was extracted from HGFs using the TRIzol reagent (Invitrogen). Subsequently, cDNA synthesis was carried out using a PrimeScript RT reagent kit (Takara Bio, Inc., Shiga Japan). Quantitative real-time PCR was performed using the cDNA as the template in a 7500 Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). PCR amplification was conducted using genespecific primers and a high-fidelity DNA polymerase, with GAPDH (encoding glyceraldehyde-3-phosphate dehydrogenase) serving as the internal control. The 2−ΔΔCt method was used to calculate the fold changes in gene expression relative to the control group [25]. For the ACTA2 (actin alpha 2, smooth muscle (α-SMA)) gene, the forward primer sequence was 5-'TGGAAAAGATCTGGCACCAC-3', and the reverse primer sequence was 5'TCCGTTAGCAAGGTCGGATG-3'.
Multipotential differentiation
The differentiation of DPSCs into multiple cell types was achieved by using osteogenic (ScienCell; Cat. no.7531) and adipogenic test kits (ScienCell; Cat. no.7541) in accordance with the manufacturer’s instructions. After the induction process, the cells were fixed using 4% paraformaldehyde and subsequently stained with alizarin red S (Sigma-Aldrich) for osteogenesis or Oil Red O (Sigma-Aldrich) for adipogenesis.
Establishment of animal models, interventions, and measurements
The study conformed to the updated ARRIVE 2.0 Guidelines. Eight-week-old male Sprague-Dawley rats were obtained from the Laboratory Animal Research Center at AFMU, following approval from the Animal Experiment Administration Committee of the University (No. 2021-010). The rats were anesthetized with a gas anesthesia machine (R540, RWD, Shenzhen, China). The rats were placed in an induction box, and anesthesia was induced with a concentration of 3-4% isoflurane. The depth of anesthesia was assessed by shaking the induction box, and if the rat was unable to turn back on its own, it was indicated that the rat was under anesthesia. The rats were removed and placed in a prone position, and anesthesia was maintained by nasal inhalation of 1-2.5% isoflurane at a gas flow rate of 0.5–0.7 L/min. At the end of the experiment, isoflurane was turned off and the rats were allowed to breathe in pure oxygen for 5–10 min to wake up. The animal model was established by local application of arecoline. Arecoline (SA9640, Solarbio, 10 mg/ mL) was first dissolved in PBS. To create an animal model, a brush with bristles made of Dupont silk (9 mm in length and 0.02 mm in diameter) was used. The brush, which had a head diameter of 6 mm, was dipped into a solution of 15 mg/mL arecoline and applied to both sides of the buccal mucosa. A pressure of 6 N was exerted on the brush in a direction perpendicular to the mucosa, while each side of the buccal mucosa was subjected to 40 strokes. Following application, the animals were fasted for two h. After 12 weeks of daily treatment, a stable white lesion developed in the buccal mucosa of the rats, indicating the presence of OSF. The OSF animal models were then randomly divided into four intervention groups: OSF, OSF + PBS, OSF + GC/PBS, and OSF + DPSC/PBS (n = 6 per group). The negative control group received PBS treatment, while the positive control group received glucocorticoids (GC/PBS, 20 mg/mL). DPSCs were resuspended in 100 µL of PBS at a concentration of 1 × 106 cells/mL. Each intervention group received weekly intra-cheek injections of 50 µL of the assigned treatment for either 4 or 8 weeks. Throughout the intervention period, weekly measurements were taken for body weight, mouth opening, and the area of the buccal lesion under isoflurane anesthesia. Measurements were carried out using procedures described previously [16]. Specifically, sulfate paper was laid over the lesion site in the oral mucosa of the rats, and the size and shape of the lesion were clearly presented on the paper. After that, the sulfate paper was removed and placed on grid paper to measure and record the specific dimensions of the lesion. All animals were kept in an isolated room with constant temperature and humidity. At the end of the intervention period, after rats were anesthetized with isoflurane inhalation, all animals were euthanized by cervical dislocation and oral mucosal tissues were collected for subsequent analysis. The time endpoint for animal modeling was 12 weeks, and the time endpoints for injection intervention were 4 and 8 weeks. All experimental procedures were carried out in accordance with the “Animal Research: Reporting of In Vivo Experiments” guidelines for preclinical animal studies.
Histological staining
Oral mucosal tissue samples were fixed in 4% paraformaldehyde for 24 h, dehydrated through a graded ethanol series, cleared with xylene, and embedded in paraffin. Section (5 μm thickness) were obtained using a microtome. Histological staining was carried out using Hematoxylin and Eosin (H&E), Masson’s Trichrome (Masson) and Sirius Red staining. Image analysis of the stained sections was conducted using ImageJ software.
Scanning electron microscopy (SEM)
The samples for SEM were initially fixed with 2.5% glutaraldehyde. Following fixation, the samples underwent a dehydration process using a series of graded ethanol concentrations. Once dehydrated, the samples were air-dried and subsequently sputtercoated with a thin layer of gold to their enhance surface conductivity, allowing for better imaging. The surface topography of the samples was visualized using a field-emission scanning electron microscope (FE-SEM, S-4800; Hitachi, Tokyo, Japan) operating at an acceleration voltage of 5 kV. To analyze the SEM images, ImageJ software was employed. This software facilitated the calculation of porosity based on the obtained SEM images.
Atomic force microscopy (AFM)
Paraffin sections were analyzed using an atomic force microscope (Keysight 5500; Keysight Technologies, Santa Rosa, CA, USA) operating in tapping mode. Images were captured using a silicon probe (PPP-NCLR-20; Nanosensors, Neuchatel, Switzerland) with a 42-N/m force constant and a 161-kHz resonance frequency. To assess mechanical properties, a probe (SD-Sphere-CONT-m-10; Nanosensors) with a 0.2-N/m force constant and a 13-kHz resonance frequency was used, achieving a 300-nm indentation depth at each section position. Six different locations within each tissue sample were measured and then averaged for analysis.
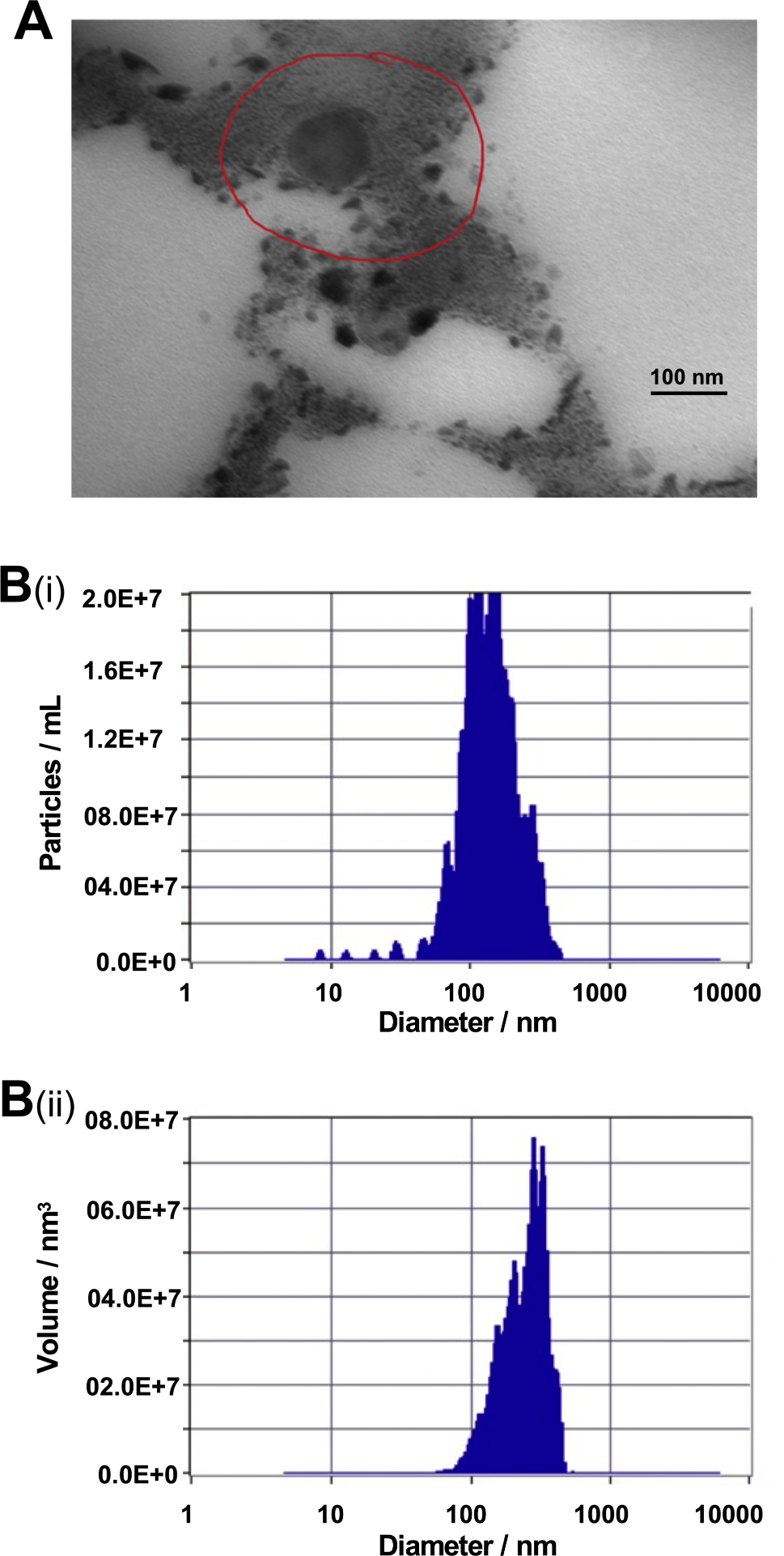
Preparation of conditioned media
DPSCs were seeded at 3 × 106/ml and when achieving 80-90% confluence, the medium was changed to a serum-free DMEM (Gibco, Rockville, MD, USA). After incubation for 48 h, the medium was collected, centrifuged at 2500 rpm for 3 min, and then filtered through 0.22-µm pore filters (Millex®-GP; Merck Millipore Ltd., Billerica, MA, USA).
Statistical analysis
The data were analyzed using GraphPad Prism 9 software (GraphPad Inc., La Jolla, CA, USA). The values are presented as the mean ± standard deviation (SD). All experiments were conducted independently at least three times. To assess the statistical significance between two groups, Student’s t-test was employed. For comparisons involving multiple groups, either one-way or two-way analysis of variance (ANOVA) was performed, followed by Tukey’s test. The significance levels were defined as *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.





留言 (0)