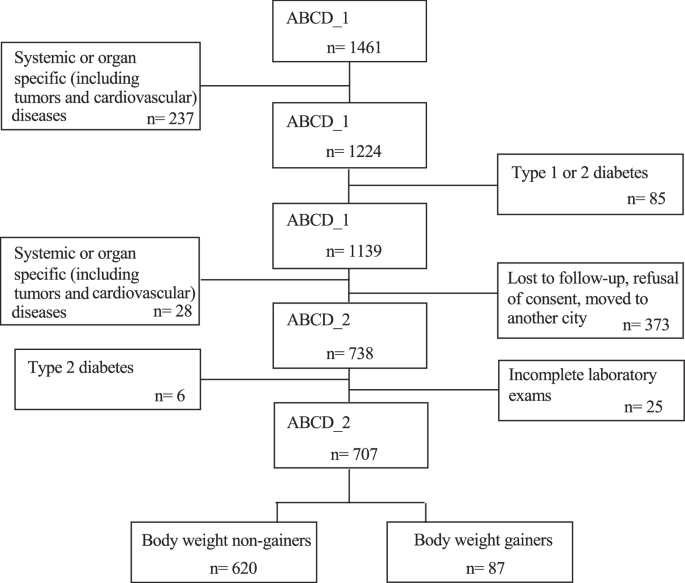
In this study, G comprised ~12% of the initial cohort; they experienced a significant increase in body weight, ~10 kg more than the NG. The G and NG groups had comparable initial body weights, BMI, body fat distributions (Table 1), and habitual energy intakes. However, as expected, 4 years later, G significantly increased their energy intake, which was higher than that of NG (Table 3), thus confirming that energy intake is the main critical factor influencing energy balance and body weight change. In general, participants in the ABCD study improved their dietary choices following initial, albeit generic, nutritional suggestions provided during the ABCD_1 study. Therefore, the macronutrient composition of the habitual diet was similar in both groups. Four years later, both groups significantly reduced their habitual fat intake and increased their carbohydrate intake (protein intake remained unchanged). However, only NG significantly reduced the glycemic index of the diet and improved adherence to the Mediterranean diet, as indicated by the MEDI-Lite score. These data also suggest that individualized prevention campaigns promoting healthy lifestyles are effective. By addressing clinical issues, providing strategies, and offering personalized suggestions, they can guide lifestyle changes in a nonintensive manner.
We previously investigated this cohort for variants in the PNPLA3 and TM6SF2 genes to identify people at a higher risk of liver steatosis (PNPLA3) and fibrosis (TM6SF2), a condition frequently associated with obesity, diabetes, and metabolic syndrome [25]. Interestingly, in this study, the G group had a significantly higher prevalence of the CG and GG variants of PNPLA3 gene than that of the NG group. Multivariate analysis revealed that the change in body weight, such as in G, was independently associated with age, changes in energy intake, and the presence of the CG or GG alleles of the PNPLA3 gene. Consistent with our findings, Cinque et al. [26] investigated liver steatosis in a small cohort of individuals during the COVID-19 lockdown and found that the PNPLA3 G allele was independently associated with body weight gain. However, to the best of our knowledge, this is the first report of an independent association between these alleles and body weight gain or obesity in a general population cohort. This association does not appear to be related to chance, as it may have a congruent biological explanation. Indeed, PNPLA3 is expressed in response to feeding and is involved in lipid autophagy (lipophagy), particularly in the liver, and to a lesser extent in adipose tissue, especially when there is an excess of dietary fatty acids [27,28,29]. Therefore, a defective protein may not ensure an efficient mechanism of autophagy that protects liver cells from excessive fat accumulation. Hence, we hypothesized that PNPLA3 might contribute to the destruction of a small but significant amount of dietary-derived fats introduced in excess, thus contributing to mitigating a positive energy balance and consequent body weight gain. If we postulate that autophagy mechanisms can eliminate as little as 5 g (or even less) of fatty acids daily, it could potentially prevent around 2.0–2.5 kg of body weight gain per year. This aligns with the amount of weight gained over 4 years by the participants in the G group of our study. However, it is essential to emphasize that this is merely a hypothesis that requires further investigation and remains speculative at this stage.
The relationship between insulin resistance (IR) and obesity has not yet been elucidated. However, our study clearly demonstrates that IR does not precede weight gain; rather, it is a consequence of weight gain. In fact, initially, the G and NG groups had the same anthropometric characteristics and did not have different values of both serum insulin concentrations and HOMA-IR. However, 4 years later, insulin levels as HOMA-IR increased significantly in the G group, which exhibited values higher than those in the NG group. As expected, the final HOMA-IR independently correlated with measures of adiposity (BMI) of central fat distribution, such as waist circumference, and, interestingly, with the amount of habitual energy intake, regardless of the type of macronutrient excess. Therefore, the higher the energy intake, the greater the IR, which supports the possibility that IR is a protective response in favor of the intracellular environment when exposed to an excess of energy substrate. If an excessive amount of glucose rapidly accumulates inside the cell, there is a risk of osmotic damage. Second, IR is a protective mechanism against oxidative damage caused by excessive nutrient and energy accumulation. Supporting this hypothesis, mitochondrial oxidative stress is known to occur due to intracellular energy substrate overflow [30]. Furthermore, a high glucose flux might force pathways, including glycogen synthesis, polyol, and hexosamine pathways, and the production of advanced glycation end-product precursors (AGEs), resulting in cellular damage [31,32,33]. Hoehn et al. [34] demonstrated in vitro and in a murine model that mitochondrial superoxide production in response to energy substrate overflow drives IR, as it is a sensor of cellular nutrient homeostasis. Therefore, IR protects the intracellular environment from oxidative stress, and mitochondrial superoxide may be a signal that drives a cellular response to dampen glucose uptake via the antagonism of GLUT4. One logical consequence is that there might be a need to reconsider the idea of pharmacologically eliminating IR. Attempting to remove this protective mechanism could have adverse effects and should be approached with caution. Similar to all compensatory mechanisms, persistent IR is expected to favor other well-known clinical problems of metabolic syndromes, such as type 2 diabetes and atherosclerosis. Therefore, the only possible rational approach to treat IR seems to be to reduce energy intake with diet, possibly using anti-obesity drugs that regulate food intake when diet alone fails.
This study had some important limitations. First, only PNPLA3 and TM6SF2 genes were evaluated, and other known genes involved in beta cell function and IR were not considered. However, these two genes were included in other investigations of liver steatosis in the ABCD study. This study did not aim to investigate the specific genetic causes of body weight gain and insulin resistance; however, we did not exclude the need for further in-depth genetic investigations of this cohort. Another important limitation is that we used HOMA-IR as a surrogate assessment of insulin resistance instead of the gold standard technique, that is, the euglycemic hyperinsulinemic clamp (EHC). However, EHC has been previously shown to correlate well with HOMA-IR values [35]; it is not a simple technique to perform in large cohorts, and to our knowledge, there is a paucity of studies using this technique beyond the first 6 months post weight loss bariatric surgery interventions [36, 37]. Unfortunately, we did not perform body composition analysis in the ABCD_1 study; therefore, we could not include body composition changes in our analysis. However, the evolution of body circumference was consistent with body weight change, and we are confident that the data on body composition modification would not have contradicted the conclusions of this study.
This study demonstrated that excessive energy intake is primarily associated with body weight gain and that genetic factors might favor a positive energy balance. Body weight gain is followed by insulin resistance, which is correlated with energy intake, and is probably a protective mechanism against intracellular oxidative stress resulting from energy substrate overflow.






留言 (0)