Peptides and drugs
PSELT, the SELENOT-mimetic peptide [SELENOT43–52 (H–Phe–Gln–Ile–Cys–Val–Ser–Sec–Gly–Tyr–Arg–OH)] and its inactive form, indicated as inert-PSELT [I-PSELT (Ser 46,49)], were chemically synthesized by automated peptide synthesizer (CEM, Saclay, France) as previously reported [19]. Dulbecco’s Modified Eagle Medium F-12 (DMEM/F-12), Dulbecco’s Modified Eagle Medium (DMEM), Dulbecco’s phosphate buffer saline (DPBS), penicillin/streptomycin (P/S), L-Glutamine, UltraPure™ DNase/RNase-Free Distilled Water, Lipofectamine® 2000 Reagent were from Thermo Fisher Scientific (Waltham, MA, USA). Foetal bovine serum (FBS) and 0.25% Trypsin–EDTA were from Corning (New York, USA). Bovine serum albumin (BSA), and non-fat dried milk were from PanReac AppliChem (Glenview, IL, USA). KCl, NaCl, NaHCO3, CaCl2, MgSO4, KH2PO4, NaH2PO4, Na2HPO4, mannitol, glucose, Na-pyruvate, β-nicotinamide adenine dinucleotide (NADH), reduced disodium salt hydrate, 2,4 dinitrophenylhydrazine (DNPH), diethyl ether, ethylenediaminetetraacetic acid (disodium salt), diethylenetriamine pentaacetic acid, pyrogallol, streptomycin sulfate, tween-20, isoproterenol, 4',6-diamidino-2-phenylindole (DAPI) were from Sigma Aldrich (Saint Louis, MO, USA). Absolute ethanol and hydrochloric acid were from Carlo Erba Reagents (Cornaredo, Milan, Italy). Before each experiment, all solutions were freshly prepared.
Animals
Experimental procedures were carried out using male healthy control Wistar (WST) rats and spontaneously hypertensive heart failure (SHHF) rats, a congenital model of dilated cardiomyopathy with hypertension progressing to HF [24] (Charles River laboratories, Milan-Italy). Animals were individually housed in cages under controlled light (12 h light/dark cycle), temperature (23–25 °C) and humidity (50–55%) and fed ad libitum with standard diet (Envigo, Udine-Italy). The study was conducted in accordance with the Declaration of Helsinki, the Italian law (D.L. 26/2014), the Guide for the Care and Use of Laboratory Animals [U.S. National Institutes of Health (NIH), Bethesda, MD, USA] and the Directive 2010/63/EU of the European Parliament on the protection of animals used for science. The project was approved by the Italian Ministry of Health, Rome, and the ethics review board.
In vivo study: experimental groups
In vivo treatments in rats started at the 15th month of age, time at which SHHF animals display significant loss of cardiac function, together with biochemical, structural, and hemodynamic alterations denoting a mid/late-stage of HF, as widely reported [24, 25]. Both WST and SHHF rats were intraperitoneally (i.p.) treated with saline (NaCl 0.9%) or PSELT (80 µg/Kg) every three days for eighteen days according to a similar protocol adopted by Yashiro et al., [26]. This dose of PSELT (corresponding to ~ 65 nmol/kg) is similar to the PSELT nanomolar concentrations able to exert cardioprotective action, as indicated in our previous publications [19, 21, 22]. Depending on saline or PSELT administration, WST and SHHF rats were divided in the following 4 groups: (i) WST + saline (WST, n = 6); (ii) WST + PSELT (WST + PSELT, n = 6); (iii) SHHF + saline (SHHF, n = 6); (iv) SHHF + PSELT (SHHF + PSELT, n = 6).
At the end of the in vivo protocols, animals were anesthetized with i.p. injection of ethyl carbamate (2 g/kg body weight), and then sacrificed. In particular, for biochemical evaluation, blood samples were collected with heparinized syringes and plasma was separated by centrifugation at 3000g (15 min, 4 °C). Plasma samples were used for the quantification of interleukin-1β (IL-1β), tumor necrosis factor-α (TNF-α), brain natriuretic peptide (BNP), and lactate dehydrogenase (LDH). Cardiac tissues were used to detect galectin-3 (GAL-3), evaluate the ultrastructure and molecular signaling, and assess performance ex vivo.
Enzyme-linked immunosorbent assays (ELISAs)
Plasma levels of IL-1β, TNF-α, and BNP were detected by ELISAs provided by Elabscience Biotechnology Inc. USA (IL-1β: E-EL-R0012; TNF-α: E-EL-R2856; BNP: E-EL-R0126) according to the manufacturer’s instructions and as previously described. Cardiac levels of GAL-3 were quantified by ELISA assay from MyBioSource, San Diego, USA (MBS761093) San Diego, United States according to the manufacturer’s instructions.
Lactate dehydrogenase (LDH) activity
The enzymatic activity of LDH in plasma samples was spectrophotometrically evaluated according to McQueen method [27] and as previously reported [28, 29]. Enzyme activity, expressed as IU/L, was determined by monitoring the absorbance decrease at 340 nm resulting from NADH oxidation.
Transmission electron microscopy (TEM) analysis on cardiac sections
After in vivo treatments, cardiac tissues were fixed in 2.5% glutaraldehyde (in 0.1 M phosphate buffer pH 7.4) for 2 h at 4 °C. Then, three washes were carried out in order to remove residual fixative, and post-fixation was performed in 1% osmium tetroxide (in 0.1 M phosphate buffer pH 7.4) for 2 h at 4 °C. Samples were washed 3 times with phosphate buffer, gradually dehydrated using increasing concentrations of acetone and then embedded in Araldite (Fluka, 10,951). Ultrathin sections by using an RMC Power Tome series ultramicrotome and a diatome diamond knife were prepared and collected on copper grids (EMS, G 300 Cu) to be examined with a Jeol JEM 1400 Plus electron microscope (Peabody, Massachusetts, USA) at 80 kV.
Western blot analysis on cardiac tissues
The apex of LV of the hearts deriving from each experimental group was homogenized in ice-cold RIPA lysis buffer supplemented with a mixture of protease and phosphatase inhibitors and centrifuged at 15,000g for 20 min at 4 °C, as detailed in previous studies [18, 19, 21]. For insoluble fraction assessment, LV was homogenized in urea-thiourea buffer [7 M urea, 2 M thiourea, and 4% (w/v) CHAPS, in 30 mM Tris–HCl (pH 8.5)] [30] supplemented with protease and phosphatase inhibitors and centrifuged at 15,000g for 15 min at 4 °C. The supernatant was collected, and the protein concentration was determined by Bradford assay. Equal amounts of proteins (30–80 μg) were separated on 10% SDS-PAGE gels for desmin and p53, on 12% SDS-PAGE gels for SELENOT, CTGF, and p21, and on 15% SDS-PAGE for phosphorylated gamma-H2A histone family member X (γH2AX, H2AX Ser139 phosphorylation) and transferred to nitrocellulose membrane by trans-blot turbo system (Bio-Rad). Membranes were blocked in 5% non-fat dried milk at room temperature for 1 h, washed three times with tris-buffered saline containing 0.1% Tween 20 (TBST) and incubated overnight at 4 °C with the specific primary antibodies diluted 1:3000 [desmin (PA5-16,705)], 1:500 [p21 (SC-6246)], 1:1000 [CTGF (TA806803), p53 (sc-126), and SELENOT (LS-C168948)] in TBST containing 5% bovine serum albumin (BSA) or TBST containing 1% non-fat dried milk (γH2AX, 05–636, final concentration 0.5 µg/ml). β-actin antibody (1:1000) (SC-47778 C4) was used as loading control. Then, membranes were incubated with peroxidase-linked secondary antibodies at room temperature for 1 h (anti-rabbit and anti-mouse diluted 1:2000 and 1:1000, respectively). Immunodetection was performed using Clarity Western ECL Substrate (Bio-rad). Bands densitometry was evaluated by measuring area and pixel intensity using ImageJ 1.6 software (National Institutes of Health, Bethesda, MD, USA), as previously indicated [18, 19, 21].
Measurement of matrix metalloproteinases (MMPs) activity by zymography
The enzymatic activity of MMPs was assessed by SDS-PAGE zymography using gelatin as substrate. An equal amount of proteins for heart samples (60 µg) was loaded under non-denaturing conditions on 8% SDS-PAGE gels containing 0.1% gelatin. Once electrophoresis was completed, the gels were washed for 30 min with buffer I [Tris–HCl (pH 7.5) and 2.5% Triton X-100], and then incubated overnight at 37 °C in buffer II [150 mM NaCl, 5 mM CaCl2, 50 mM Tris–HCl (pH 7.6)]. Finally, the gels were stained with 2% Coomassie Brilliant Blue R250 (Sigma Aldrich), 25% methanol, 10% acetic acid and then de-stained in 2% methanol–4% acetic acid for 1 h [31]. The gelatin digestion areas by MMPs were visualized and quantified with ImageJ 1.6 software. MMP-2 activity was expressed as a percentage of total band area in the 4 experimental groups.
Ex vivo study: Langendorff isolated rat heart perfusion
At the end of in vivo procedures and after anaesthesia, chest was opened and heart excised in order to be connected to the Langendorff apparatus under a retrograde perfusion with Krebs–Henseleit (KH) solution at a constant flow rate of 12 mL/min, 37 °C, as reported in previous studies [18, 19, 21, 32, 33]. Hemodynamic parameters were recorded every 10 min and analysed using PowerLab data acquisition system (AD Instruments, Sydney, New South Wales, Australia).
Basal conditions
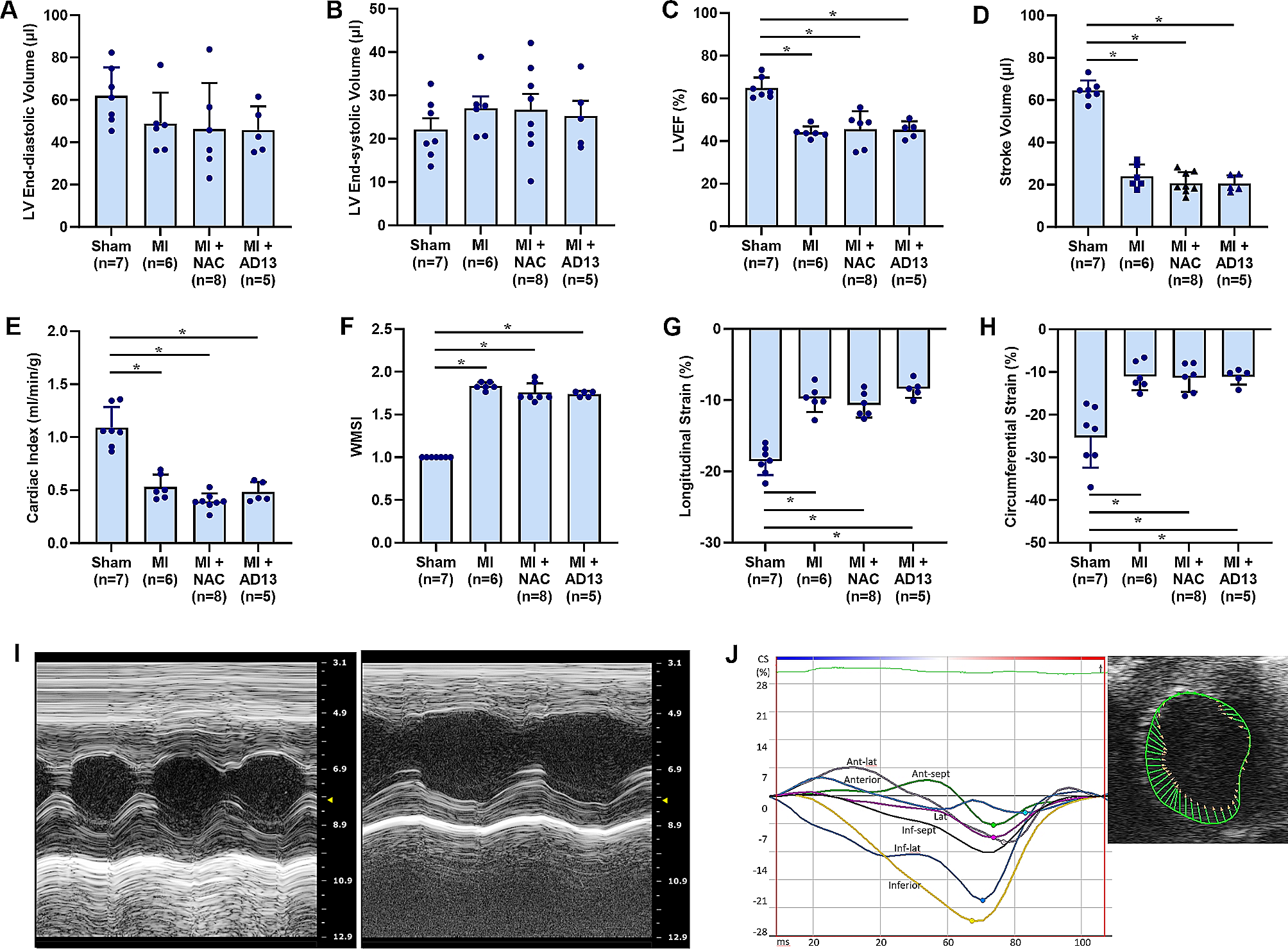
Basal cardiac performance was evaluated by monitoring inotropism, in terms of developed left ventricle (LV) pressure (dLVP; mmHg, index of contractile activity) and maximal value of the first LVP derivative [+ (LVdP/dT) max; in mmHg/s, index of maximal LV contraction rate], lusitropism, assessed through the maximal rate of LVP decline [− (LVdP/dT) max; mmHg/s], and LV end diastolic pressure (LVEDP; mmHg), coronary vasomotility, assessed by monitoring coronary pressure (CP; mmHg), and heart rate (HR) changes (beats/min) used to evaluate chronotropism.
Ischemia/Reperfusion (I/R) protocols
Hearts of each experimental group were subjected to I/R protocols in order to reproduce I/R injury (IRI). After 40 min stabilization, hearts were subjected to 30 min, zero-flow global ischaemia followed by 120 min of reperfusion. Post-ischemic cardiac recovery was assessed by monitoring post-ischemic dLVP and LVEDP, being this latter an index of contracture, defined as its increase of 4 mmHg above the baseline level [18, 19, 21, 34, 35]. The stability of all preparations was evaluated by measuring cardiac performance every 10 min and these parameters were stable up to 190 min. Accordingly, sham hearts were perfused with KH buffer for 190 min.
Infarct size (IS) evaluation
Immediately after IRI protocols, hearts were rapidly removed from the perfusion apparatus to assess infarct area by nitro-blue tetrazolium staining in a blinded fashion manner, as detailed in our previous studies [18, 19, 21]. Unstained necrotic tissues were carefully separated from stained viable tissues to calculate IS, expressed as a percentage of total LV mass.
In vitro study in murine macrophages and rat and human cardiomyocytes: cell cultures
Murine macrophages RAW 264.7 cell line (ATCC, Cat# TIB-71) were cultured in DMEM (4.5 g/L glucose) (Gibco) containing 10% FBS, 2 mM glutamine, 1% P/S and incubated at 37 °C in a humidified chamber, 5% CO2. For experiments RAW 264.7 cells were seeded in 6-well plates for 24 h in a humidified atmosphere, 5% CO2 at 37 °C. H9c2 cardiomyoblast cells (ATCC, Cat# CRL-1446) were cultured in DMEM/F-12 (Gibco) supplemented with 10% FBS, 1% P/S (Thermo Fisher Scientific) and incubated at 37 °C in a humidified chamber containing 5% CO2. For experiments cells were seeded in complete medium and incubated for 48 h at 37 °C, 5% CO2, as previously reported [21, 22, 36]. AC16 human cardiomyocytes (Millipore-Sigma Aldrich, Cat. # SCC109) were grown in DMEM/F-12 supplemented with 12.5% FBS, 2 mM glutamine and 1% P/S, and incubated in a humidified atmosphere of 95% air and 5% CO2 at 37 °C. For experiments, cells were plated in complete medium for 24 h at 37 °C, 5% CO2, as previously reported [37, 38].
Analysis of CD80 by flow cytometry in RAW 264.7 macrophages
RAW 264.7 cells were seeded 6 well plates and treated with vehicle (saline, NaCl 0.9%) (CTRL) or LPS (100 ng/ml) [39, 40] and PSELT (5 nM) alone or in co-treatment for 24 h. At the end of the treatments, cells were washed with cold DPBS detached with trypsin–EDTA and centrifuged. The pellet was resuspended in 100 µl of cold DPBS containing FITC anti-CD80 (B7-1) monoclonal antibody (11–0801-82) (Thermo Fisher Scientific) according to the manufacturer's instructions. After 30 min incubation at 4 °C, RAW 264.7 cells were washed with DPBS (1X) and centrifuged at 500 g for 5 min, then re-suspended in DPBS and analyzed by flow cytometry (CytoFLEX Beckman, Beckman Coulter, Milan, Italy). Data analysis was performed using CytExpert Beckman Coulter software (Beckman Coulter, Milan, Italy).
Morphological staining in H9c2 cardiomyoblast cells
Morphological changes were assessed by May-Grunwald Giemsa (MGG) staining, as previously reported [36, 41]. H9c2 cells were seeded in 60 mm culture dishes, treated with vehicle (saline, NaCl 0.9%), isoproterenol (ISO) (100 µM) [42], ISO + PSELT (5 nM) or PSELT alone [22], while another set of H9c2 cells were treated with vehicle, ISO, ISO + I-PSELT (5 nM) or I-PSELT alone, and incubated in a humidified atmosphere at 37 °C for 48 h. After MGG staining, H9c2 cardiomyocytes were visualized by using Olympus BX41 microscope, and the images were taken with CSV1.14 software, using a CAM XC-30 for image acquisition. Cell-surface area was analyzed by using Image J 1.6 (NIH). Data were expressed as percentage of a relative increase in cell-surface area.
Gene silencing of endogenous SELENOT by small interfering RNA (siRNA) in H9c2 cardiomyocytes
Gene silencing for SELENOT was performed in H9c2 cardiomyocytes, as previously described [21, 22]. Briefly, cells were seeded in 60 mm cell culture dishes and incubated for 48 h at 37 °C, 5% CO2. SELENOT siRNA (100 nM) was transfected into H9c2 cells using Lipofectamine 2000 reagent following the manufacturer’s instructions (Invitrogen). Negative control si-RNA (si-NC) was used to evaluate sequence-specific silencing from non-specific effects. Both control siRNA-A and siRNA for SELENOT were purchased from Santa Cruz Biotechnology. H9c2 cardiomyocytes were transfected in serum-free medium for 6 h; then, medium was replaced with full media and cells were incubated for 36 h at 37 °C, 5% CO2. Cells were treated with vehicle or ISO (100 µM) for 48 h and, at the end of the treatments, cell morphology was evaluated by MGG staining, as described above. Data were expressed as percentage of relative increase in cell-surface area.
Western blot analysis on H9c2 cardiomyocytes
H9c2 cells treated with vehicle, ISO (100 μM), ISO + PSELT (5 nM) and PSELT alone were processed as reported previously [22, 36, 41]. After establishing protein concentration in the supernatant by Bradford assay, 50 μg of proteins were loaded on SDS-PAGE gel, then electroblotted onto a nitrocellulose membrane and probed with the primary specific antibody against SELENOT. Antibody against β-actin was used as loading control. Following the incubation with the primary antibodies, membranes were incubated with peroxidase-conjugated secondary antibodies at room temperature for 1 h (anti-rabbit and anti-mouse diluted 1:2000 and 1:1000, respectively). Immunodetection and densitometric analysis were performed as described above.
Gene expression assessment by qPCR
H9c2 and AC16 cardiomyocytes were grown in 6-well plates to reach 70–80% confluence and treated with vehicle (CTRL), ISO (100 μM), ISO + PSELT, and PSELT (5 nM) alone for 48 h. At the end of treatments, total RNA was isolated using TRIzol reagent (Invitrogen) according to the manufacturer’s protocol. 2 μg of total RNA were reverse transcribed to provide cDNA using High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems™, Thermo Fisher Scientific). cDNA was amplified by qPCR using SYBR™ Select Master Mix (Thermo Fisher Scientific) according to the manufacturer’s instructions on QuantStudio™ 1 Real-Time PCR System apparatus (Thermo Fisher Scientific). Samples were analyzed in duplicate (n = 3 independent experiments). 18S rRNA was used as an internal control. The primers used for the amplification were listed in Table 1. Relative gene expression levels were calculated using the 2−ΔΔCt method [43, 44].
Table 1 Primer sequences used for qPCR amplificationAssessment of cell-surface area in AC16 human cardiomyocytes
Cellular hypertrophy in AC16 cardiomyocytes was evaluated by Alexa Fluor 568–conjugated phalloidin (Thermo Fisher Scientific) following the manufacturer's instructions (Thermo Fisher Scientific). In brief, cells were seeded on coverslip and incubated for 24 h in humidified chamber 37 °C, 5% CO2 and then exposed to vehicle (CTRL), ISO (100 µM), ISO + PSELT, PSELT (5 nM) for 48 h. After treatments, cells were fixed with 3.7% paraformaldehyde in DPBS for 10 min at RT, then permeabilized with 0.1% Triton-X100 for 5 min and incubated with 1% BSA in DPBS for 30 min [45]. Cells were stained with Alexa Fluor 568–conjugated phalloidin for 20 min and then nuclei were counterstained with DAPI. Images were taken CSV1.14 software, using a CAM XC-30 for image acquisition. The cell surface area from 3 independent fields was measured with ImageJ software.
Quantitative detection of intracellular and extracellular levels of SELENOT in AC16 human cardiomyocytes
AC16 cells were treated with vehicle (CTRL), ISO (100 μM), ISO + PSELT, and PSELT (5 nM) alone for 24, 48, 72, and 96 h. At the end of each treatment, intra-cardiomyocyte and extracellular levels of SELENOT were quantified in cell extracts and cell supernatants, respectively, by using a “Human Selenoprotein T ELISA” sandwich kit provided by MyBioSource (Cat. No MBS166821) according to the manufacturer’s instructions.
Transmission electron microscopy (TEM) analysis on human cardiomyocytes
AC16 human cardiomyocytes exposed to vehicle (CTRL), isoproterenol (ISO) (100 µM), ISO + PSELT (5 nM), PSELT for 48 h, were fixed with 3% glutaraldehyde in 0.1 M phosphate buffer overnight at 4 °C. Post-fixation proceeded in buffered osmium tetroxide, followed by dehydration in a graded acetone series, as previously described [22]. Ultrathin Sects. (60–90 nm in thickness) were cut with a diamond knife, mounted on copper grids (G300 Cu), and imaged using a Jeol JEM 1400-Plus electron microscope operating at 80 kV.
Statistical analysis
Data were expressed as means ± SEM. A two-way ANOVA and nonparametric Bonferroni’s multiple comparison test (for post-hoc ANOVA comparisons) were used for hemodynamic analyses. A one-way ANOVA and nonparametric Newman–Keuls multiple comparison test (for post- hoc ANOVA comparisons), were used for all the other analyses. Values for *p < 0.05, **p < 0.01, ***p < 0.001 were considered statistically significant. Statistical analysis was conducted using Prism 5 (GraphPad Software, La Jolla, CA, USA).


留言 (0)