Inborn Errors of Immunity (IEIs) encompass a group of genetically and phenotypically heterogeneous inherited disorders that impede the development and/or function of the human immune system [1]. Primary Immune Regulatory Disorders (PIRDs) are linked to autoimmunity, autoinflammation, and/or disruptions in lymphocyte homeostasis [4]. Commonly, defects in T cells and their tolerance induction, B cells, immunoglobulins, and class-switch recombination, as well as genes affecting multiple cellular subsets, constitute the prevalent issues predisposing IEI patients to autoimmunity [19]. The advent of NGS technologies has revealed an expanding list of monogenic defects underlying IEIs, with diagnostic yields ranging from 15 to 79% [13].
In these 40 index patients, a total of 38 disease-causing variants (16 of which were novel) were identified. Among these, 23 variants were deemed likely pathogenic or pathogenic, while 16 were classified as variants of unknown significance (VUS). Out of the 38 different PIRD mutations, 22 were missense, eight were nonsense, five were in/dels, and one represented a single-base substitution in an exon–intron junction sequence, likely affecting a splice site.
This study marks the first instance of using next-generation sequencing within our country to investigate the distribution of mutations in the PIRD patient population. Turkey has reported a notably high rate of consanguineous marriages [20]. This phenomenon has contributed to a heightened prevalence of autosomal recessive inherited diseases, including Inborn Errors of Immunity. Among our cohort, recessive PIRD genes constituted 45% of cases, while dominant variants accounted for 40%, and X-linked PIRD genes constituted 15%.
In a Dutch cohort, NGS-based assessment for IEIs showed the highest yields among pediatric patients, within the immune dysregulation cluster, most patients received a diagnosis of familial hemophagocytic lymphohistiocytosis (HLH), with additional cases including autoimmune lymphoproliferative syndrome (ALPS), primarily attributed to pathogenic FAS variants [14]. Conversely, our cohort is primarily clustered within the immune dysregulation category, with a diagnosis of syndromes characterized by autoimmunity.
In a study from India, diseases involving immune dysregulation were observed in 20 patients. Most frequently, most of the patients are diagnosed with FHL [21].
In another study from Egypt, genetic assessments were conducted for 39 patients exhibiting immune dysregulation disorders. Among them, 21 individuals from 15 distinct consanguineous families displayed variations in the LRBA gene. Other infrequent genetic diagnoses included variants in IL10RA, IL10RB, FOXP3, AIRE, DOCK8, SLC7A7, UNC13D, PRKCD, SH2D1A, RIPK1, and FAS variants [22].
Our immune gene panel differs from the Dutch, Iranian, and Egyptian cohorts, as it involves a distinct panel lacking certain HLH genes found in the Primary Immune Deficiency Research Panel v2. Our clinicians preferred the HLH panel over the immune panel for FHL, which resulted in relatively lower FHL findings when assessed with our immune panel.
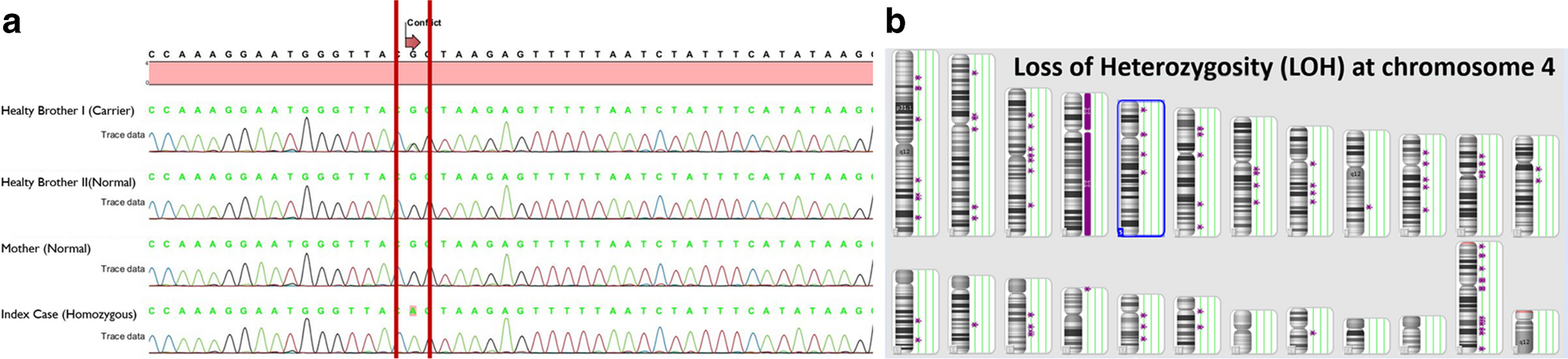
LRBA deficiency is an autosomal recessive disorder arising from biallelic mutations in the LRBA gene (OMIM #614,700). Clinically, it is characterized by early-onset hypogammaglobulinemia, autoimmune manifestations, susceptibility to inflammatory bowel disease, and recurrent infections. While partial isodisomy-associated LRBA deficiency has been reported previously [23], our study reports the first instance of LRBA mutation becoming homozygous through whole chromosome uniparental disomy (UPD).
Homozygous AIRE mutations c.769C > T (Arg257Ter), c.415C > T (p.Arg139Ter), and c.254A > G (p.Tyr85Cys) exhibit a founder effect in the Finnish, Sardinian, and Iranian Jewish populations, respectively [24]. The c.769C > T (Arg257Ter) variant in exon 6 has been identified in 89% of Finnish APECED alleles but is also the most prevalent across other ethnic groups. A literature review involving 23 published Turkish APECED patients revealed that the Finnish major mutation, c.769C > T (Arg257Ter), is prevalent in the Turkish population [24]. Of significance, three out of four (75%) APECED cases in our study featured the c.769C > T (Arg257Ter) variation.
Currently, there are nine genes associated with IEIs in which mutations have been detected for both loss-of-function and gain-of-function mutations: CFB, C3, CARD11, STAT1, STAT3, WAS, JAK1, IFIH1, and ZAP70 [2]. Gain-of-function (GOF) mutations bestow a different function upon the mutant gene due to the mutation it undergoes, leading to unexpected protein production. This type of mutation increases the transcription of a gene, endowing it with heightened activity and mobility, often referred to as a hypermorphic gene. Conversely, loss-of-function (LOF) mutations render the gene product dysfunctional. A gene product completely devoid of function is termed a null allele or amorphous allele. If the mutant type retains partial function, it is referred to as a hypomorphic allele. For instance, while functional mutations in STAT3 may manifest with lymphoproliferation, including lymphadenopathy and hepatosplenomegaly, and early-onset multisystem autoimmunity, STAT3 loss-of-function mutations underlie hyperimmunoglobulin E syndrome (Job’s syndrome). This syndrome is characterized by recurrent infections, eczema-like skin rashes, and vulnerability to severe lung infections. While both LOF and GOF of STAT3 cause immune deficiency, GOF leads to infections distinct from those observed with LOF, accompanied by more common connective tissue abnormalities [25, 26]. Moreover, a 5-year-old male with a novel ZAP70 c.1448C > T (p.Ser483Phe) variant underwent hematopoietic stem cell transplantation (HSCT), yielding successful clinical and immunologic outcomes.
CTLA4 deficiency is a rare disorder profoundly disrupting immune system regulation, leading to conditions such as intestinal disease, respiratory infections, autoimmune issues, and enlarged lymph nodes, liver, and spleen. Abatacept offers a potentially effective treatment for patients with documented CTLA-4 deficiency, inducing and sustaining remission of enteropathy [27].
In summary, PIRDs can manifest similar clinical profiles despite distinct genetic defects, and conversely, the same genetic defect can result in diverse clinical presentations. Beyond clinical diagnosis, identifying the molecular defect causing the disease is pivotal for prognosis prediction, treatment planning (e.g., abatacept, HSCT), preimplantation genetics, prenatal diagnosis, and carrier identification. Despite the genetic diversity underpinning PIRDs, genetic counseling assumes a crucial role in managing and shaping future decisions for affected families.
In conclusion, the integration of NGS into the study of PIRD molecular genetics provides a comprehensive and nuanced understanding of the disorder. From identifying rare variants to uncovering novel mutations, NGS emerges as a cornerstone technology, offering unprecedented insights that pave the way for personalized medicine and improved patient outcomes in the realm of Inborn Errors of Immunity. A key conclusion from this multicenter study: This report represents the inaugural utilization of NGS for diagnosing the Turkish PIRD cohort, offering novel insights that expand the spectrum of clinical manifestations attributed to various PIRD-related mutations.



留言 (0)