Data collection
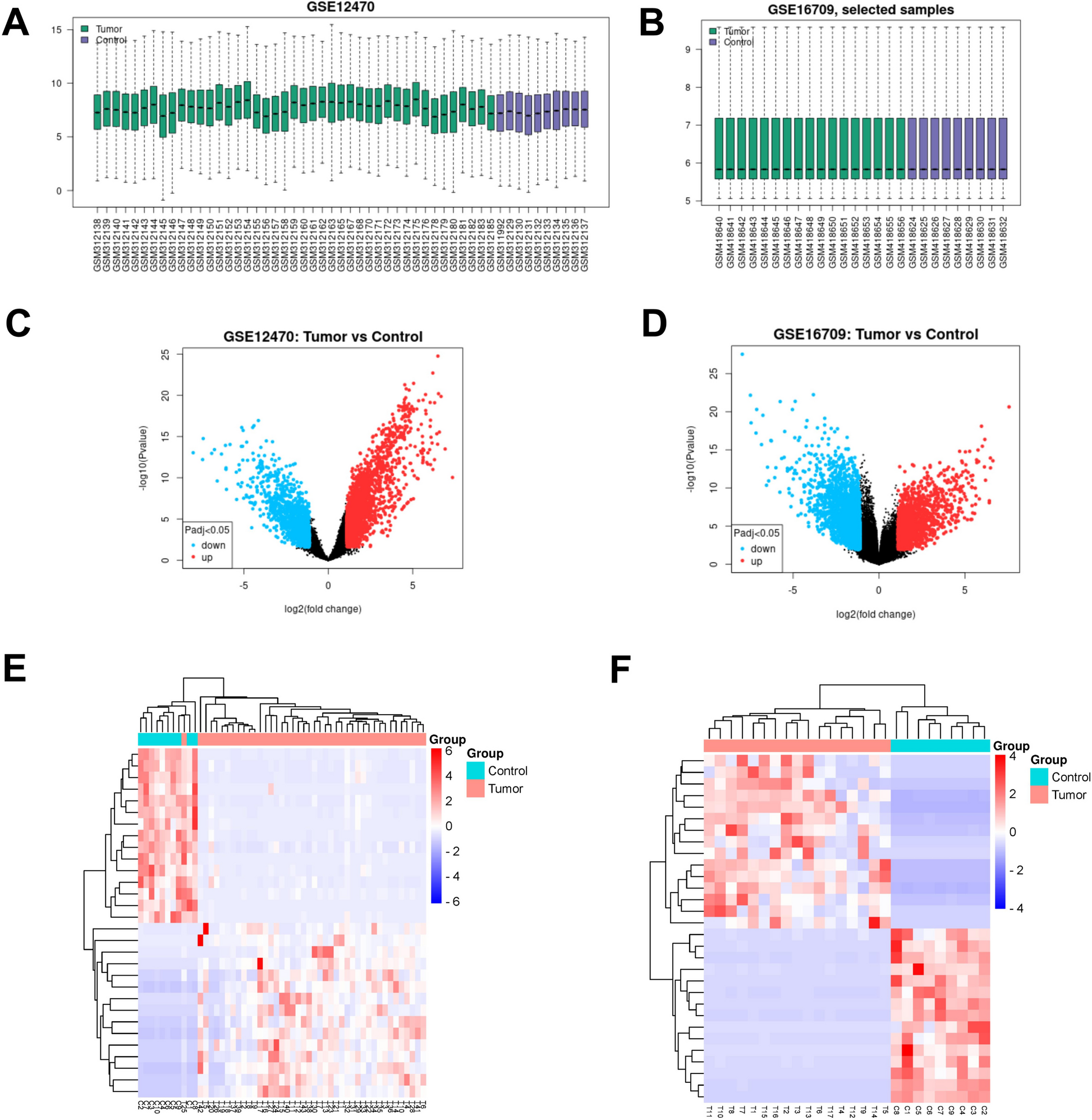
The RNA sequences of 422 patients (50 normal tissues and 374 tumor tissues) were extracted from the TCGA-HCC database, and the RNA sequences of 445 patients (202 normal tissues and 243 tumor tissues) were extracted from the ICGC-LIRI-JP database. Ferroptosis-related genes were acquired from FerrDb, an online source that supplies a comprehensive and updated checklist of ferroptosis markers, regulative molecules, and ferroptosis-disease associations [12]. In general, we identified 175 (Driver:76; suppressor:47; marker:75) ferroptosis-related genes (Table S1). Pearson correlation evaluation was used to identify the relationships between ferroptosis-related genes and lncRNAs. Connections with the value of correlation coefficients |R|> 0.5 and a P-value < 0.001 were considered statistically substantial. The clinicopathological attributes of HCC people, consisting of age, gender, stage, TMN, survival condition, and survival time, were accumulated. In this study, we identified significant differentially expressed lncRNAs (DElncRNAs) between HCC tumors and normal liver tissue (absolute log2FC > = 1 and FDR < 0.05). After screening differentially expressed genetics (DEGs), we performed functional enrichment evaluation and KEGG analysis using upregulated and downregulated ferroptosis-related differentially expressed genetics (DEGs). Enrichment analysis using metascape for EDGs data.
Building of the ferroptosis-related lncRNAs prognostic signature
Univariate Cox regression, LASSO, and multivariate Cox regression evaluations were used to establish the last variables for building the prognostic threat score model. stratified based on risk rating (Coefficient lncRNA1 × expression of lncRNA1) + (Coefficient lncRNA2 × expression of lncRNA2) + ⋯ + (Coefficient lncRNAn × expression lncRNAn). Based on the threat scores for OS, HCC clients with fibrosis were separated into high- or low-risk groups using the typical score as a cut-off, and also Kaplan–Meier contours were calculated for both teams.
Predictive nomogram building and validation
To execute genetics established enrichment evaluations stabilized, all genetics matters were imported right into the GSEA software application as well as the Molecular Signature Database (MSigDB) Trademark, KEGG, and also Genetics Ontology gene libraries were utilized to identify enriched gene sets [13], which were then searched in the TCGA-HCC database. Enriched gene sets were selected based on statistical significance (incorrect exploration rate FDR q value < 0.25 and normalized p-value < 0.05) [14]. We built a nomogram incorporating typical medical variables such as age, gender, grade, tumor stage, and the risk rating originated from the prognostic trademark to assess the possible 3- and 5-year OS of clients with HCC.
Gene expression and immunity analysis
At the same time, the CIBERSORT [15, 16], ESTIMATE [17], quanTIseq [18], xCell [19], MCP-counter (or mMCP-counter for mouse) [20], EPIC [21], and TIMER [22] formulas were analyzed, the CC or cell sorts of immune reactions in heterogeneous examples among risky as well as low-risk teams based on ferroptosis-related lncRNAs trademark. The differences in immune feedback under various formulas were revealed using a Heatmap. In enhancement, ssGSEA was utilized to quantify the tumor-infiltrating immune cell subgroups between both groups and assess their immune function. Many possible immune check factor molecules were determined that could be targeted and incorporated with potential medication therapy [23].
Cell culture and transfection
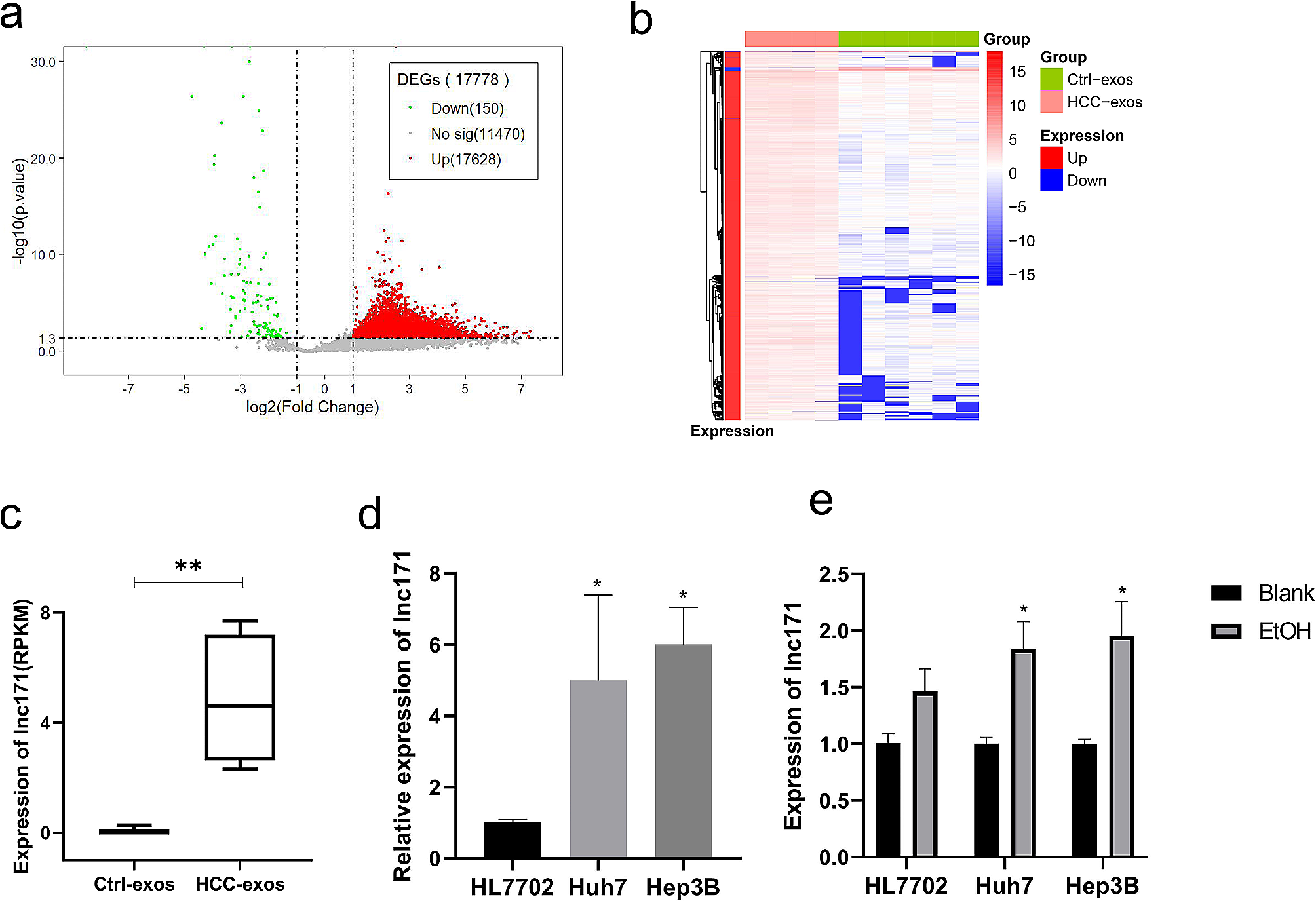
Liver cancer cell lines (HuH-7, SKhep1, hep3B, hepG2) were ordered from the Chinese Academy of Sciences (Shanghai, China) and cultured in DMEM medium containing 25 mM glucose, 4 mM L-glutamine and 1 mM pyruvate (Gibco, 12800). All cell lines were authenticated using short tandem repeat (STR) profiling (by GENEWIZ Co. Ltd. at Suzhou, China). After authentication, large frozen stocks were made for future use. All cell lines were used within 15 passages (less than 2 months)after reviving from the frozen stocks. The medium was supplemented with 10% FBS and 1% penicillin/streptomycin. Cells were kept at 37 °C in a humidified incubator with 5% CO2. The trypan blue exclusion assay was used to detect cell growth. All cell lines were tested for mycoplasma contamination, and no cell lines were contaminated. The miR-375-3P mimic, inhibitor, and corresponding control oligonucleotides were purchased from Shangya (Fuzhou, China). The transfection of miR-375-3P mimics, inhibitors, or corresponding NCs into cells was performed using Lipofectamine 3000 (Invitrogen). The lentiviruses of Flag-SRSF5, NRAV, overexpression vector (vector), sh-NRAV, and empty vector (sh-NC) were constructed by Genechem (Shanghai, China). The cells were transfected with lentivirus according to the manufacturer's instructions. The sequences of shRNA against specific targets are available in Table S2.
Nucleocytoplasmic separation
Total RNAs were extracted from cell lines and tissues using Trizol (Invitrogen, USA). The RNA of nuclear and cytoplasmic was separated and extracted using PARIS Kit (Life technologies, USA) according to the manufacturer’s instructions. The expression levels of NRAV, U6, and GAPDH genes in various RNA samples obtained by qRT-PCR were measured.
Tissue specimens
Eighty-six matched HCC tissues and paracancerous tissues were obtained from patients with HCC diagnosed by The First Affiliated Hospital of Zhengzhou University. All experiments involving human samples and clinical data were approved by the Accreditation Committee of The First Affiliated Hospital of Zhengzhou University.
RNA extraction and quantitative real-time PCR (qRT-PCR) analysis
According to the manufacturer's protocol, total RNA from HCC cells, tissues, and matched non-cancerous tissues was isolated using the TRIzol Reagent (Thermo Fisher Scientific, Waltham, MA, USA). Reverse transcription was performed using the PrimeScript RT Reagent Kit (Takara, Dalian, China). Bulge-loop™ miRNA RT-qPCR Primers were applied to determine the level of miRNAs. The real-time PCR reactions were performed using StepOnePlus™ Real-Time PCR System (Thermo Fisher Scientific, MA, US). The program settings on temperature cycling were followed as instructed by the manufacturer. GAPDH was the cytoplasmic control, and U6 was the nuclear control. The sequences of primers are listed in Table S2.
Western blot analysis
In brief, proteins were isolated from HCC cells using RIPA buffer (Solarbio, Beijing, China) supplemented with proteinase inhibitors, and the protein concentration was determined with a BCA reagent (Beyotime, Beijing, China). Cell lysates were separated on SDS–polyacrylamide gels and then transferred onto polyvinylidene difluoride (PVDF) membranes (Millipore). After the membranes were blocked in 5% skim powdered milk for 2 h, they were incubated with primary antibodies overnight at 4 °C. The primary antibodies used in this study included: ACSL4 (HUABIO, ET7111-43), GPX4 (HUABIO, ET1706-45), SLC7A11 (Abcam, ab175186), and GAPDH (Proteintech, 80,570–1-RR), FLAG(Sigma-Aldrich, No. F7425). Next, the membranes were incubated with secondary antibodies (HUABIO, Hangzhou, China) at room temperature for 1 h. After washing three times, the targeted proteins were visualized using enhanced chemiluminescence (ECL) reagent (Millipore, MA, USA). GAPDH was used as the loading control in this study. Flag-SRSF5 served as a positive control.
Colony formation assay
Cells were seeded into 12-well plates at an initial density of 700 cells/well and cultured for 2 weeks. The colonies were fixed in 4% paraformaldehyde and stained with 0.1% crystal violet (Sigma, St. Louis, MO, USA). The visible colonies were counted using a light microscope. The number of colonies in triplicate wells were measured for each treatment group.
Transwell migration assays
Transwell assays were conducted using 24-well plates with chamber inserts with 8.0 μm pores (Corning). The cells were digested 24 h after transfection and resuspended in a serum-free medium. Then, 3 × 104 cells per well were placed into the upper chambers. μlLike a cell nutritional attractant, the lower chamber was filled with a 500 μl medium with 10% FBS. After incubation at 37 °C for 24 h, cells in the upper chamber were gently removed with a cotton swab. The migrated or invaded cells were fixed with 4% polyformaldehyde and visualized by staining with crystal violet for 20 min. Last, the migrated cells were imaged and quantified by capturing five randomly chosen microscopic fields using an inverted microscope (Olympus, Japan). All experiments were performed in triplicate.
Cell proliferation assays
For the Cell Counting Kit-8 (CCK-8) assay, the treated HCC cells were seeded into 96-well plates at a concentration of 3 × 103 /well. Then, 10 μl of CCK-8 assay solution (Dojindo, Japan) was added and incubated in the dark for 2 h. The absorbance at 450 nm was measured every 24 h with a microplate reader (BioTek Instruments, USA). All experiments were repeated three times.
RNA immunoprecipitation (RIP)
Following the manufacturer's protocol, RNA immunoprecipitation (RIP) assay was performed using Magna RIP™ RNA-binding protein immunoprecipitation kit (Millipore, Billerica, MA, USA). The cell extract was incubated with magnetic beads conjugated with anti-Argonaute 2 (AGO2) or anti-IgG antibody (Millipore, Billerica, MA, USA) for 6 h at 4 °C. The beads were washed and incubated with Proteinase K to remove proteins. Finally, isolated RNA was extracted using TRIzol Reagent (Thermo Fisher Scientific, Waltham, MA, USA), then the purified RNA was subjected to qRT-PCR analysis. The specific binding sites of NRAV and miR-375-3P were determined by annolnc2 (http://annolnc.gao-lab.org).
Dual-luciferase reporter assay
The full-length wild-type (WT) sequence of NRAV and the indicated mutant NRAV containing the predicted miR-375-3P binding sites were separately synthesized and cloned into the dual-luciferase reporter vector (Genechem, Shanghai, China). The resulting dual-luciferase reporter plasmids (WT or Mut) were co-transfected with the miR-375-3P mimic or inhibitor into hepG2 or HuH-7 cells, respectively, using Lipofectamine 3000. After 48 h of incubation, the relative firefly luciferase activities concerning the corresponding Renilla luciferase activities were measured and analyzed using a Dual-Luciferase Assay System (Promega, Fitchburg, WI, USA) following the manufacturer's protocol.
Iron assay
Intracellular ferrous iron (Fe2+) level was determined using the iron assay kit (ab83366, Abcam) according to the manufacturer's instructions. Cells were collected and washed in ice-cold PBS, homogenized in 5 × volumes of iron assay buffer on ice, then centrifuged (13,000 × g, 10 min) at 4 °C to remove insoluble material. The supernatant was collected, and an iron reducer was added to each sample before mixing and incubating at room temperature for 30 min. Then, 100 μl of the iron probe was added to each sample, mixing and incubating the reaction for 1 h at room temperature in the dark. The absorbance at 593 nm was measured immediately using a colorimetric microplate reader.
Lipid ROS measurement
Lipid ROS level was analyzed by flow cytometry using BODIPY-C11 (GLPBIO, GC40165) dye. Cells were seeded at 2.5 × 105 per well in a six-well dish and grown overnight for 12 h. Cells were washed once with PBS. Cells were then stained with 2 ml medium containing 5 µM of BODIPY-C11 and incubated at 37 °C for 20 min in the dark. Cells were washed twice with PBS to remove excess labeling mixture, followed by resuspending in 500 μl fresh PBS (DPBS, Gibco). The cell suspension was filtered through a 0.4 μM cell filter and subjected to flow cytometric analysis to detect the amount of intracellular lipid ROS. The fluorescence intensities of cells per sample were determined by flow cytometry using the BD FACSAria cytometer (BD Biosciences). A minimum of 10,000 cells was analyzed for each sample. Data analysis was evaluated using the FlowJo Software.
Subcutaneous xenograft model
For the subcutaneous xenograft mouse model, 10 4- to 6-week-old male athymic BALB/c nude mice (Shanghai SLAC Laboratory Animal Co,Ltd., China) were housed and fed in standard pathogen-free conditions. The male nude mice were randomly divided into two groups (n = 5 in each group) and inoculated with cells as follows: sh-NC stable transfected hepG2 Cell (2 × 106 cells); sh-NRAV stable transfected hepG2 Cell (2 × 106 cells); Cells were mixed with matrigel (1:2) and inoculated subcutaneously at the right rear back region. Tumors were measured weekly with calipers, and tumor volume was calculated as = (L × W2)/2. Five weeks later, the mice were euthanized and the tumors were isolated for further study. If a tumor grew to 2000 mm3, the experiment was terminated in advance. After measurement, the mice were euthanized with an overdose of 10% pentobarbital sodium (100 mg/kg; intraperitoneal injection), and death was confirmed by the disappearance of the heartbeat. All in vivo animal experiments were approved by the Committee on the Ethics of Animal Experiments at the Shanghai University of Traditional Chinese Medicine.
Statistical analysis
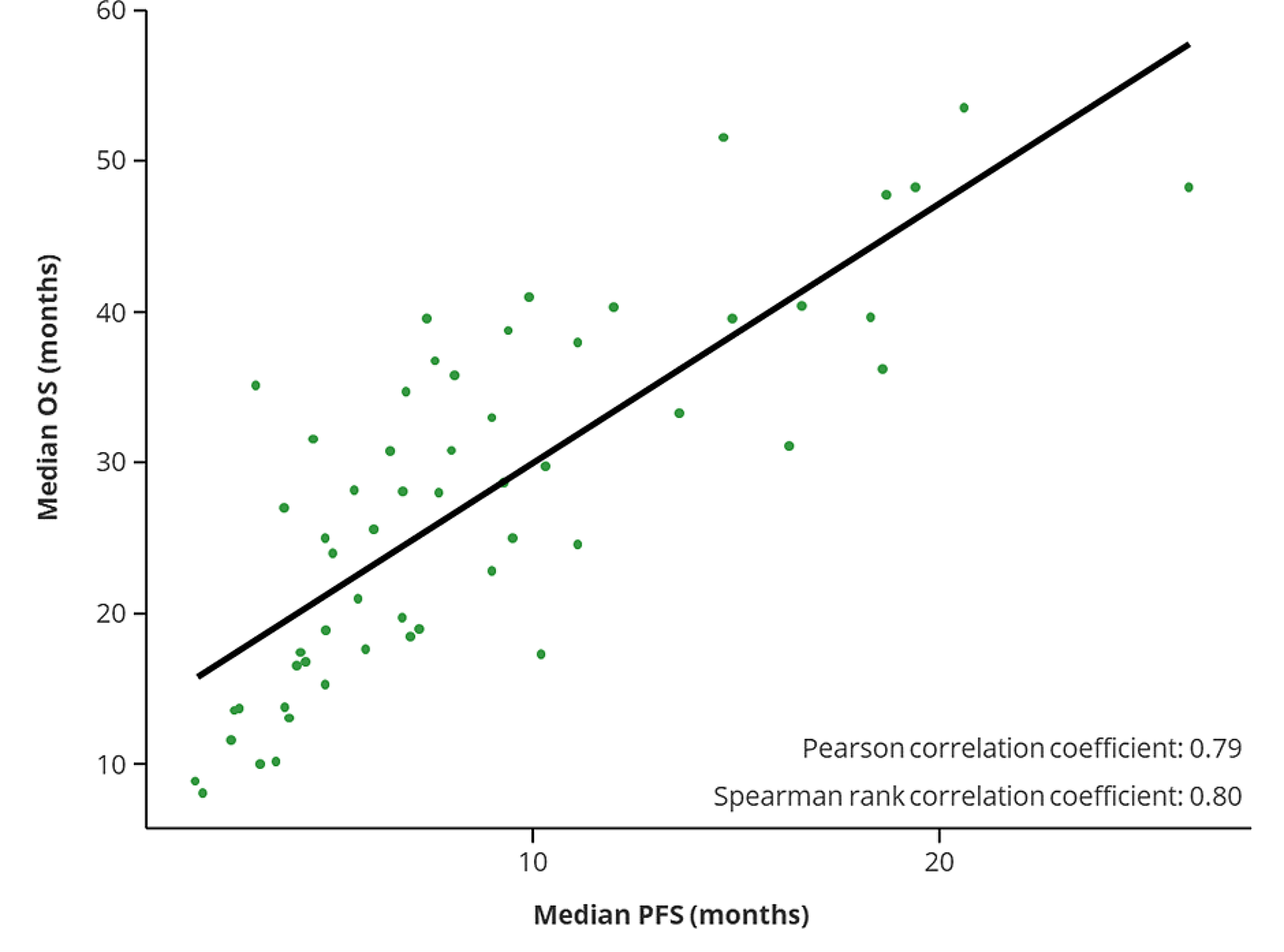
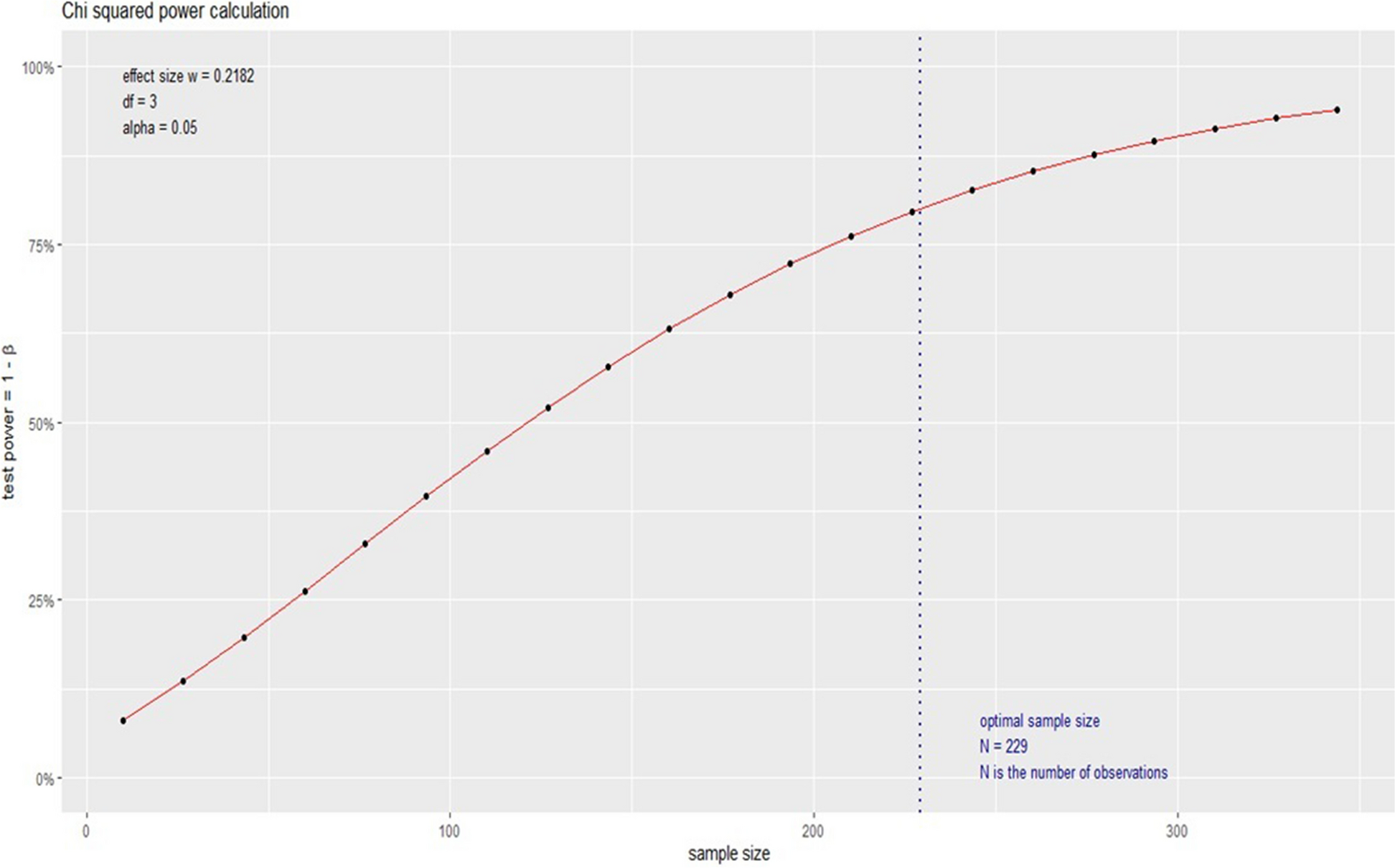
The experimental data were analyzed using statistical analysis software, including GraphPad Prism 8.0 software (GraphPad) and the R package (V.3.3.4). Differences between the indicated groups were compared using the Student's t-test and one-way analysis of variance (ANOVA) followed by Fisher's least significant difference (LSD) test. Correlations were evaluated by Pearson correlation analysis. The cumulative overall survival (OS) and progression-free survival (PFS) rates were calculated using the Kaplan–Meier method, and significance was evaluated with the log-rank test. A P-value < 0.05 was considered to indicate a statistically significant result.



留言 (0)