
記住我






The cultivated strawberry (Fragaria ×ananassa) is among the most economically important fruit crops in the world. According to the Food and Agricultural Organization of the United Nations, during 2021, more than nine million tonnes of strawberries were produced from 389,665 ha around the world (FAO, 2023). The cultivated strawberry as it is today resulted from a series of interspecific hybridizations, with the final cross between octoploid progenitors occurring only approximately 300 years ago (Edger et al., 2019; Whitaker et al., 2020). Evolutionary analysis of Fragaria ×ananassa suggested genomic contributions of four diploid progenitor species: Fragaria vesca, Fragaria iinumae, Fragaria viridis, and Fragaria nipponica (Edger et al., 2019; 2020). However, analysis from other groups has suggested that both F. viridis and F. nipponica are not among the diploid progenitors (Liston et al., 2020; Jin et al., 2023; Lyu et al., 2023; Session and Rokhsar, 2023). As identification of the progenitor species may enable greater prediction of polyploid responses to environmental stress and climate change (Liston et al., 2020), further analysis of the evolutionary history of strawberry will be necessary to confirm the identities of the diploid progenitor species.
Fragaria ×ananassa is a highly heterozygous, allo-octoploid species (2n = 8x = 56) with a phased genome length of approximately 780 Mb (Hardigan et al., 2021a; Han et al., 2022). Due to the complex nature of the allo-octoploid genome and high genomic heterozygosity, trait discovery and gene functionalization studies in strawberry are commonly performed in the diploid woodland strawberry, Fragaria vesca. Fragaria vesca is used as a model system for strawberry research for several reasons: it has a small genome size of approximately 240Mb, has a short life cycle, is relatively responsive to transformation, and is easy to propagate using both seeds and runner cuttings (Oosumi et al., 2006). Fragaria vesca is additionally the closest relative of the transcriptionally dominant diploid subgenome of Fragaria ×ananassa (Hardigan et al., 2021b), further lending to its value as a model system for strawberry research. While trait discovery and gene functional studies performed in Fragaria vesca are useful for inferring mechanisms of commercially valuable traits in the cultivated strawberry, there are also drawbacks to relying on the diploid system. The largest drawback to working with the diploid strawberry is that Fragaria vesca tends to be more homozygous at a given locus as compared to Fragaria ×ananassa. Martín-Pizarro et al. (2019) identified one such instance of this when they identified at least five alleles of the FaTM6 locus across the four homoeologous chromosomes of octoploid strawberry and only a single homozygous FvTM6 locus in the diploid strawberry. Due to the differences in heterozygosity, it can be difficult to determine which allele(s) in Fragaria ×ananassa contribute to a phenotype using data from Fragaria vesca alone, hindering attempts at genomics-assisted improvement of commercially relevant varieties.
Recent advancements in genome sequencing technology and gene annotation have enabled the assembly of numerous strawberry genomes encompassing a range of ploidy levels (Qiao et al., 2021; Han et al., 2022; Song et al., 2023a). Enabled by these genome assemblies, CRISPR/Cas genome editing is increasingly applied for the study and improvement of key strawberry traits. While not currently widespread due to several challenges, genome editing in allo-octoploid strawberry offers a unique opportunity to precisely modify traits of interest. Alternatively, tools such as RNAi, stable or transient overexpression of transgenes have been utilized to validate gene function and support identification of candidate genes. Plant breeding has also made use of genomics-assisted techniques such as marker-assisted selection and genomic selection for varietal improvement, enabled by new methods for QTL discovery and analysis.
This review covers the genomic tools and technologies available for genomics-assisted improvement of cultivated strawberries, in addition to multi-omics technologies and their applications in identifying candidate QTLs and genes. Reports of successful CRISPR/Cas genome editing for traits associated with strawberry fruit quality and production are discussed herein. In addition to these reports, this review suggests additional candidates for genome editing based on analyses in strawberry and other crops.
2 Advanced genomic resources in strawberryGenomic complexity of the polyploid genome is one of the major challenges of genome editing in octoploid strawberry. The first high quality, chromosome-scale reference genome of Fragaria ×ananassa cv. ‘Camarosa’ was developed in 2019 (Edger et al., 2019). Before the release of the first octoploid strawberry genome, there were few genome assemblies available for polyploid species, as the homoeologous nature of the subgenomes made assembly of sequences difficult (Wang et al., 2023b). As such, molecular genetic analysis of Fragaria ×ananassa prior to 2019 relied on early genome assemblies of Fragaria vesca, the first of which was published in 2011 (Shulaev et al., 2011). Recent rapid advancements in genome sequencing technology have resulted in the release of additional annotated octoploid genome sequences of increasing quality (Table 1) (Wang et al., 2023b). Advancements in genome sequencing in strawberry have not been limited only to the octoploid, as new high-quality genomes have also recently been released for the diploid model species, Fragaria vesca (Alger et al., 2021; Joldersma et al., 2022). As improvements to genome sequencing technology continue, it will be possible to better understand genomic complexity in not only octoploid strawberry, but in other polyploids as well.
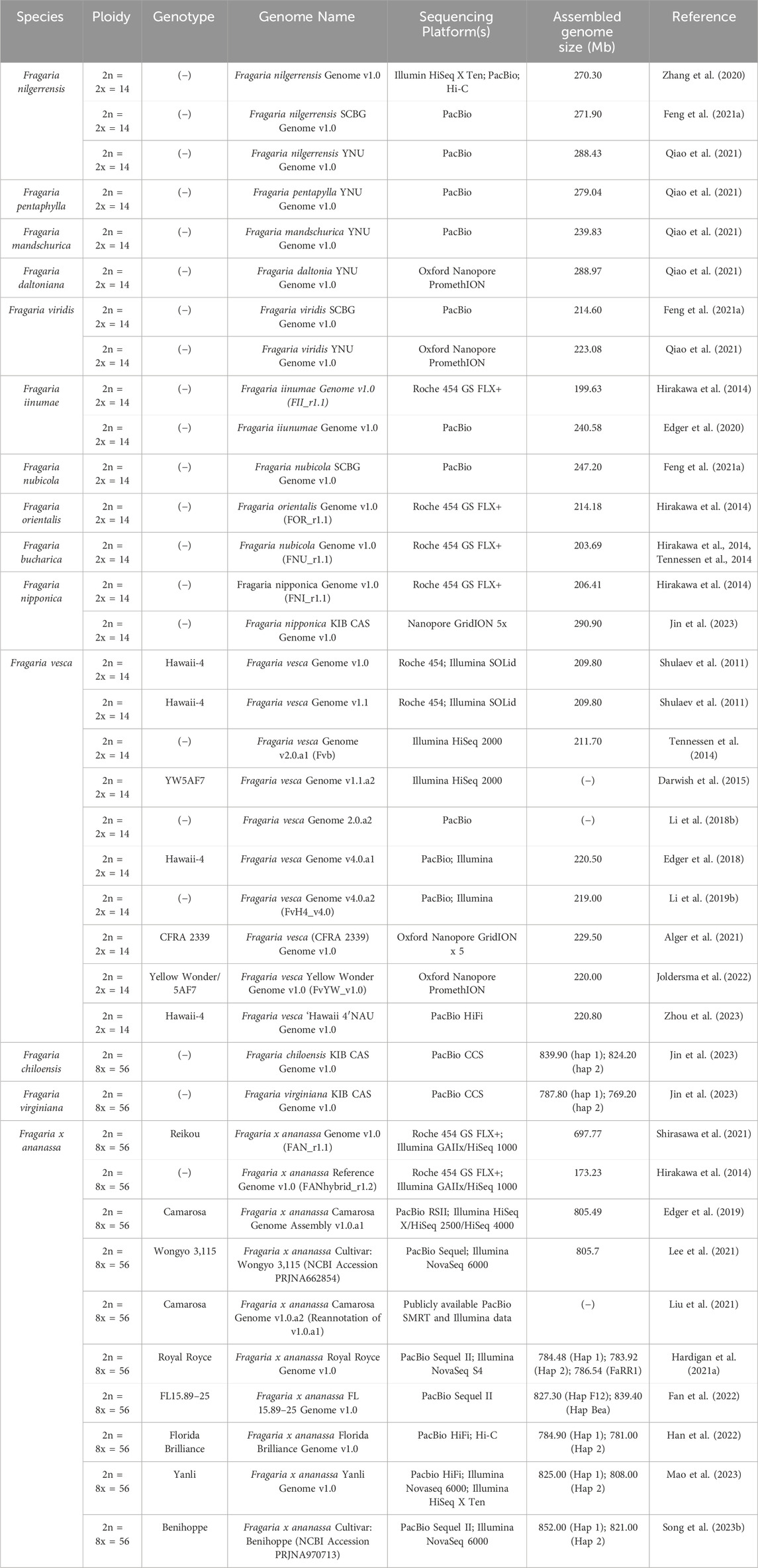
Table 1. Compilation of publicly available genome assemblies for various Fragaria species. (−) indicates value not reported.
Recently, multiple single nucleotide polymorphism (SNP) arrays for octoploid strawberry have been developed to assist with quantitative trait loci (QTL) discovery, including 50K and 90K arrays (Bassil et al., 2015; Verma et al., 2017). Using these tools to identify SNPs correlated to QTLs enables the development of molecular markers which can be utilized in breeding (Jung et al., 2017). Marker assisted selection (MAS) is a method of precision genomics-assisted breeding which relies on the implementation of genetic markers and trait associations to inform selection (Collard and Mackill, 2008). Large numbers of marker-trait associations have been generated through QTL mapping studies (Collard and Mackill, 2008; Rey-Serra et al., 2021) which makes MAS a powerful tool for precision breeding. Implementation of high-throughput assays such as high-resolution melting (HRM) and simple sequence repeat analysis (SSR) paired with DNA markers and rapid high-throughput DNA extraction methods have enabled rapid improvement of octoploid strawberry varieties at the University of Florida (Noh et al., 2017). This success highlights the significant potential of MAS for varietal improvement of fruit crops.
Genomic selection (GS) is a method of selection which utilizes genome-wide variation and phenotypic data to predict phenotypes of an unobserved population (Bernardo, 1994; Meuwissen et al., 2001; Goddard and Hayes, 2007; Montesinos-López et al., 2021). GS offers the potential for increased genetic gain within a breeding program, as it enables increased selection intensity, selection accuracy, and reduction of the generational interval (Werner et al., 2023). Additionally, GS enables faster selection of clonally propagated crops such as those in the Rosaceae family by predicting their performance as clones while they are still in the seedling stage, allowing for earlier analysis of traits that would otherwise require further physiological development, such as fruit flavor and shelf life (Werner et al., 2023). Due to the complexity of the polyploid genome, generation of genome-wide SNP arrays can be complicated, contributing to the delay in adoption of GS methods for breeding of polyploid crops (Zingaretti et al., 2019). Despite the challenges posed by genomic complexity of polyploids, the continuous release of high-quality reference genomes has allowed for the parallel improvement of SNP genotyping methods. To aid in adoption of GS in polyploids, software such as polyploid Sequence Based Virtual Breeding (pSBVB) has been developed to simulate and evaluate GS strategies in polyploids and is equipped to simulate differences between allo- and autopolyploids (Zingaretti et al., 2019). Numerous reports have now been made for successful application of GS in octoploid strawberry (Gezan et al., 2017; Whitaker et al., 2017; Osorio et al., 2021). In a study by Yamamoto et al. (2021), 105 inbred strawberry lines were developed and used to train a GS model based on phenotyping data for petiole length, leaf area, Brix, fruit firmness, and pericarp color. Using the model to predict phenotypic values for a F1 hybrid testing population derived from these 105 lines revealed that phenotypic data collected from the parental inbred lines was sufficient to predict the F1 hybrid phenotypes when the model accuracy in cross-validation is sufficient (Yamamoto et al., 2021). Pincot et al. (2020) applied GS to strawberry to evaluate improve Verticillium wilt resistance. While the inclusion of wild genotypes in the training population reduced accuracy, the results suggested a strong potential for GS to identify superior resistant individuals if the model was sufficiently trained (Pincot et al., 2020).
Due to the wide range of research in strawberry, the volume of available genomic resources has increased significantly. As such, the Genome Database for Rosaceae (GDR) was created to house a wide range of tools and data. Among these tools is a compendium of strawberry DNA tests which can be downloaded from GDR1 and implemented in breeding programs (Oh et al., 2019).
2.1 Multi-omics approaches for trait discovery for improving strawberryEnabled by the release of high-quality genome assemblies, genomic approaches have been applied to help identify candidate genomic regions and genes for several key strawberry traits, including flavor (Oh et al., 2021; Fan et al., 2022; 2023), disease resistance (Mangandi et al., 2017; Nellist et al., 2019; Nelson et al., 2021; Salinas et al., 2019), fruit firmness (Lee et al., 2021), fruit quality (Verma et al., 2017), and fruit shape (Nagamatsu et al., 2021). As genomic technologies continue to improve, they will allow for greater understanding of the genetic interactions and mechanisms underlying traits of breeding interest. QTL mapping has been performed in strawberry to identify loci underlying major quality and production traits such as day-neutrality, runner production, disease resistance, and fruit quality traits, among many others (Weebadde et al., 2008; Castro et al., 2015; Cockerton et al., 2018; Hossain et al., 2019; Alarfaj et al., 2021). Similarly, genome-wide association studies (GWAS) have been applied in strawberry for the discovery of numerous major traits (Pincot et al., 2018; Wada et al., 2020; Saiga et al., 2022). Recent advancements in genome sequencing technology have also enabled the analysis of large populations and generation of pangenomes, from which structural variants associated with key traits can be identified (Bohra et al., 2020). Pangenomics has been applied in strawberry to identify patterns in fruit color (Qiao et al., 2021). Analysis of strawberry pangenomes also resulted in interesting findings about strawberry evolution and domestication. Based on transposable element analysis of a pangenome constructed from 10 high-quality strawberry genomes, Lyu et al. (2023) suggested that Fragaria viridis may not be one of the diploid Fragaria ×ananassa ancestors as previously thought. Additionally, Qiao et al. (2021) discovered a new diploid strawberry species during assembly of their own pangenome. Discovery of transposable elements and other structural variants can be difficult when performing analysis of single genomes, however, such variants have been known to impact major agronomic traits (Tao et al., 2019), and are easier to identify through pangenome analysis. Thus, continued exploration of strawberry pangenomes may yield further insight into other major fruit quality and production traits and may assist in unravelling the complex evolutionary history of cultivated strawberry. Genomics-assisted technologies have also been widely implemented for trait discovery in crops outside of Rosaceae. New methods of QTL mapping have demonstrated capacity to resolve QTL candidates within a window of only a few kilobases (Bohra, 2013; Varshney et al., 2014; Zhang et al., 2019; Bohra et al., 2020), and in some cases, were able to generate QTL regions of which the resolution is comparable to the outcomes of sequence-based GWAS (Zhang et al., 2019; Bohra et al., 2020). Implementation of these new techniques in strawberry may enable further trait discovery for key agronomic traits.
In addition to the application of genomics for candidate gene identification, transcriptomics has been applied in strawberry to map expression quantitative trait loci (eQTLs) related to various fruit traits, including flavor (Sánchez-Sevilla et al., 2014; Barbey et al., 2020; 2021; Fan et al., 2022) disease resistance (Barbey et al., 2019), ripening and softening, and several others (Barbey et al., 2020). Strawberry transcriptome data has also recently been used to explore major postharvest issues, such as host responses to Botrytis cinerea infection (Yu et al., 2021) and differences in shelf life between cultivars (Min et al., 2020). In both cases, several candidate genes were identified which may be involved in plant defense, regulation of senescence, and shelf life (Min et al., 2020; Yu et al., 2021). Transcriptomic analyses of strawberry resulted in updates to existing genome annotations (Liu et al., 2021) and new transcriptome assemblies (Sánchez-Sevilla et al., 2017). Sufficient strawberry transcriptomic data has even been generated to allow for a meta-analysis of fruit ripening which resulted in the identification of previously unrevealed differentially expressed genes (DEGs) (Yi et al., 2021). Additionally, it is possible to identify allelic contributions based on relative expression patterns using transcriptomic data (Castillejo et al., 2020; Chandra et al., 2021; Oh et al., 2021). Considering this, transcriptomics could be a powerful tool for identifying candidates in polyploid crops such as strawberry, where unraveling the allelic contribution to a specific trait across homoeologous chromosomes remains challenging. As such, identification of transcriptionally dominant alleles may inform targeted trait improvement.
Despite the advancements in genome sequencing and transcriptomics which facilitate genome editing in octoploid strawberry, gene functional studies still rely heavily on transgenic approaches including RNA interference (RNAi) and overexpression (OE). Numerous traits, including several of postharvest interest, have been explored through transient and transgenic approaches in strawberry (Table 2). RNAi is an efficient tool for validating gene function through post-transcriptional gene silencing induced by double stranded RNA (Singh and Roychoudhury, 2023). RNAi and antisense approaches have also informed successful CRISPR/Cas strategies in strawberry (García-Gago et al., 2009; Paniagua et al., 2020; Paniagua et al., 2022; López-Casado et al., 2023). Transgenic application of tools like RNAi, antisense downregulation, and OE can be used to identify candidate genes prior to genome editing. Such transgenic lines depend on the continued expression of the recombinant constructs, which can vary depending on environmental or developmental effects. As such, these transgenic lines are not ideal to facilitate varietal improvement. Instead, identification and functionalization of candidate genes can be used to inform subsequent CRISPR/Cas genome editing. With the correct strategy, candidate genes can be knocked in or out, and alleles can be swapped for superior versions, allowing for altered gene expression or function and resulting phenotypes. The corresponding mutants can then be used in breeding schemes to further improve elite varieties. As such, transgenic approaches can be important tools to inform genome editing strategies for improvement of strawberry.
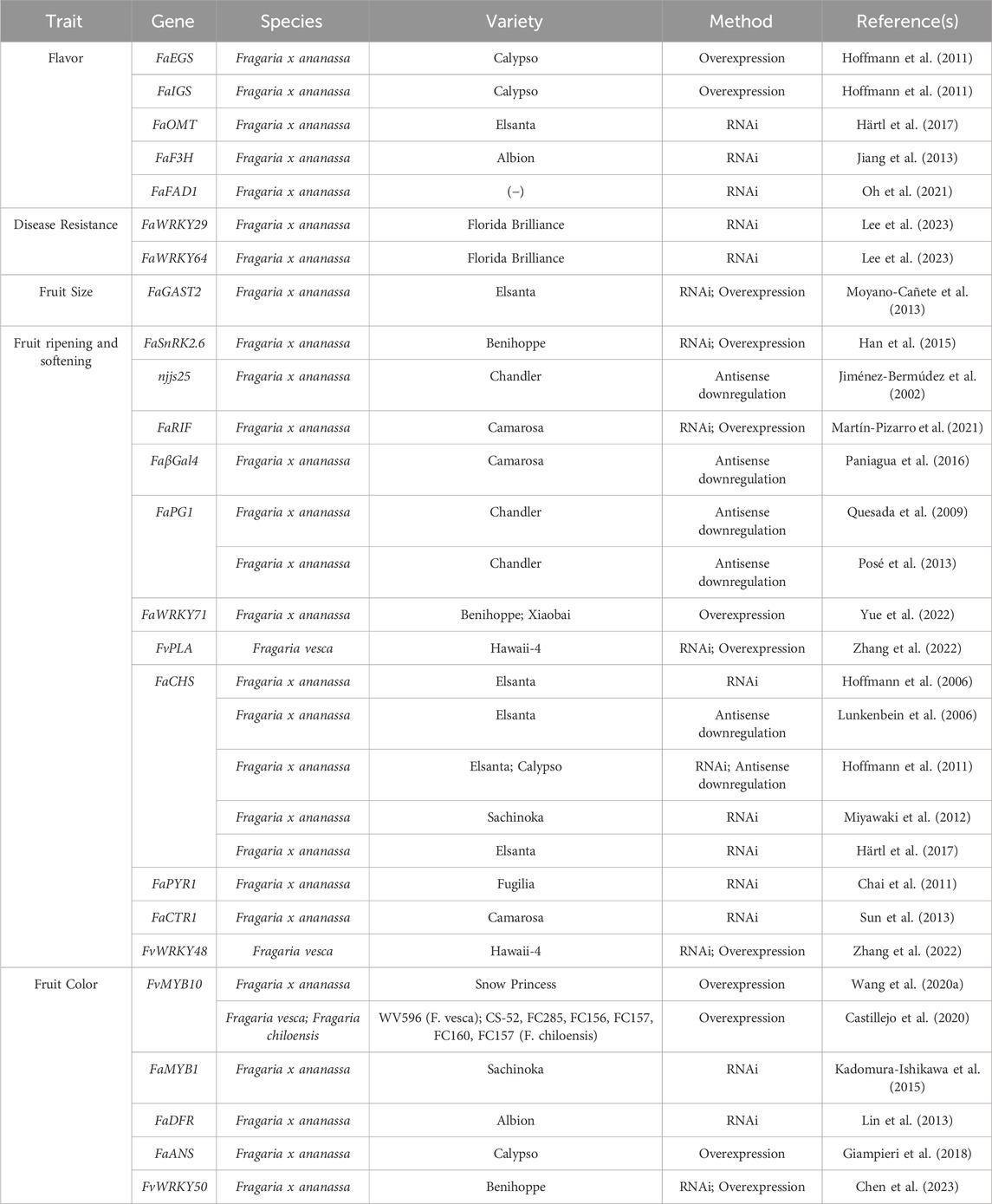
Table 2. Reported applications of transgenic approaches in strawberry for analysis of fruit quality and postharvest traits. (−) indicates value not reported.
Since metabolites are major contributors to fruit flavor and quality, it is necessary to identify genes associated with their production. Metabolomics can supplement transcriptomic and genomic data to support gene discovery on a biochemical level. In strawberry, metabolomics is frequently used in flavor studies (Schwab et al., 2008; Chambers et al., 2014; Fan et al., 2022), however, metabolomics has also been implemented in studies of plant stress response (Antunes et al., 2019), fruit development and ripening (Vallarino et al., 2018), and response to blue light (Chen et al., 2020). Like metabolomics, proteomics can be used in tandem with transcriptomic and genomic data to facilitate gene discovery. Application of proteomics and transcriptomics has been used to analyze postharvest quality changes during storage under different conditions, including controlled ozone treatments (Chen et al., 2019) and temperature stress (Lv et al., 2022). In both cases, comparisons of the differentially expressed proteins (DEPs) with the expression patterns of their respective genes found that proteosome responses mimicked the changes in postharvest quality, further supporting their proposed roles in stress response (Chen et al., 2019; Lv et al., 2022).
Multi-omics has been employed for trait discovery in strawberry and other crop species. In tomato, the correlation of SNPs, transcripts, and metabolites was used to identify new genes and pathways dictating major fruit traits (Zhu et al., 2018; Gaston et al., 2020). Similar application of multi-omics analysis in strawberry may discover novel pathways and gene candidates supporting fruit quality and production. If diverse evidence points to major genes for a desirable trait, CRISPR/Cas-mediated editing of the major gene or its repressor should have a high potential for crop improvement. As such, continual integration of genomics, transcriptomics, metabolomics, and proteomics is critical to inform CRISPR/Cas9 approaches for improvement of strawberry.
3 Genetic transformation and regeneration of strawberryFollowing trait discovery, genetic transformation can be performed to validate candidate gene function (Figure 1). While CRISPR/Cas-mediated genome editing has yet to be widely implemented and optimized for Fragaria species, numerous reports of transgenic modifications have been made. Agrobacterium-mediated transformation is currently the most applied transformation method for strawberry. Protocols using agrobacterium have undergone significant optimization to improve transformation efficiency. Thus far, protocols have been established for Agrobacterium-mediated transformation and subsequent regeneration of a range of tissues (Li et al., 2018a; Feng et al., 2019; Feng et al., 2021b; Wilson et al., 2019; Duan et al., 2021; Mao et al., 2022; Yan et al., 2023). In strawberry, agrobacterium-mediated transformation most commonly uses Agrobacterium tumefaciens strains LBA4404, GV3101, and MP90. Leaves are the most common explant material (Table 3). Transient methods of agrobacterium-mediated transformation have also been developed for fruit (Carvalho et al., 2016; Dai et al., 2020; Zeng et al., 2021; Mao et al., 2022; Lee et al., 2023) to study a range of mechanisms and traits, as well as to analyze the performance of DNA constructs prior to stable transformation. Particle bombardment (Agius et al., 2005) and protoplast transfection (Gou et al., 2020) have also been applied in strawberry for transient analyses. However, despite the demonstrated success of both methods in such analyses, stable transformation of strawberry with these approaches remains a challenge.
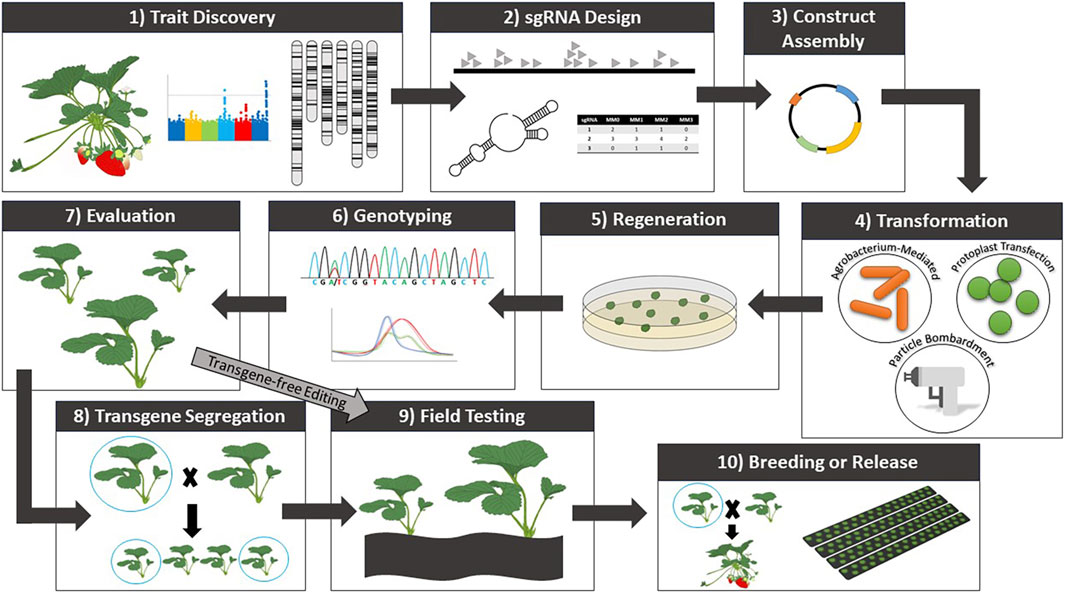
Figure 1. General workflow for trait discovery and CRISPR/Cas-mediated genome editing in strawberry.
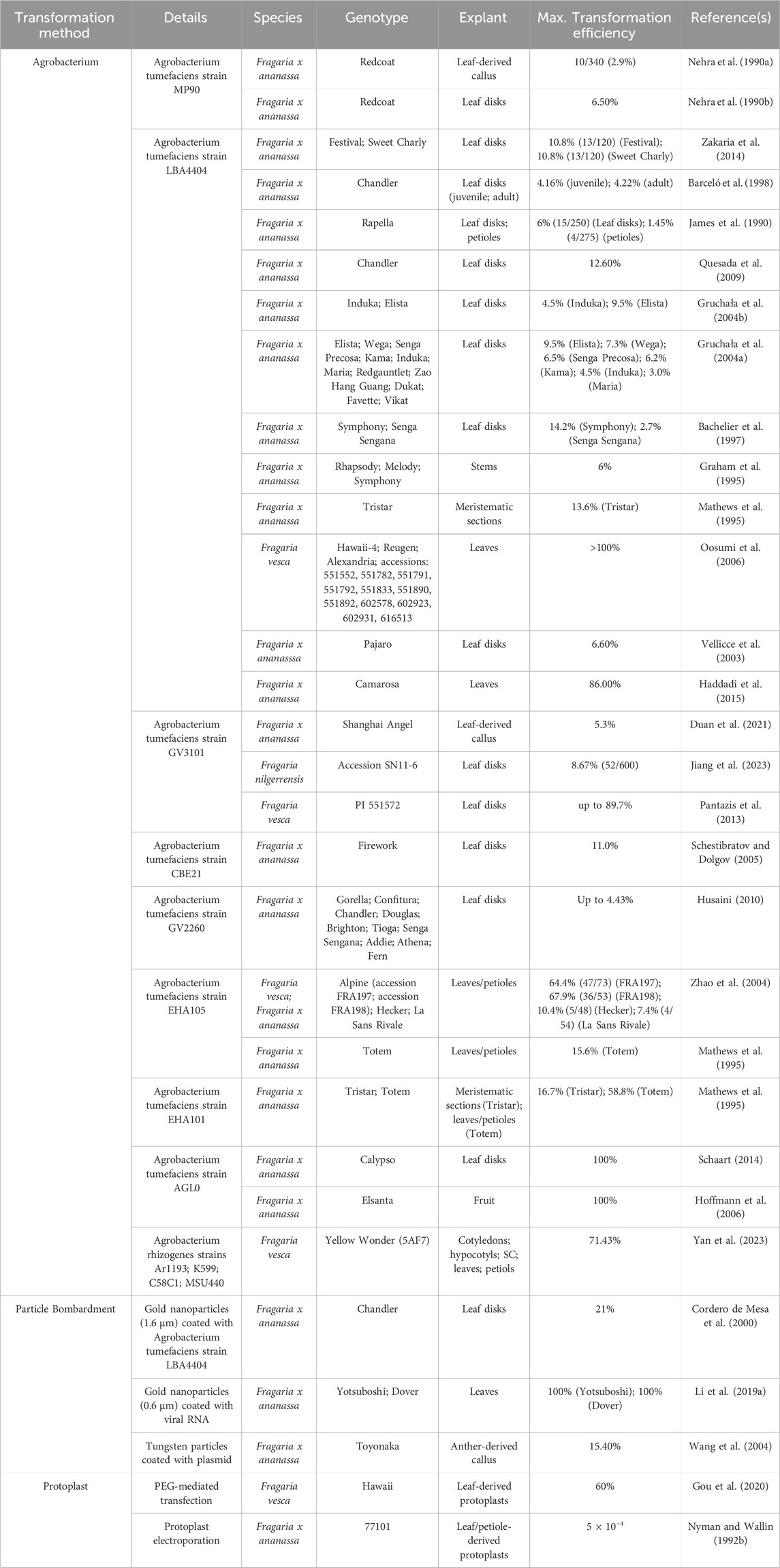
Table 3. Reported methods for genetic transformation of strawberry.
Transformation of strawberry has also been performed using the hairy root system (Yan et al., 2023). Hairy roots are valuable for functional analysis of root traits as well as for validation of transgenic and genome editing methods due to the relatively short period of root development (Ozyigit et al., 2013). Several advancements have been made to Agrobacterium-mediated transformation technologies, including improved ternary systems (Anand et al., 2018), auxotrophy of various amino acids (Aliu et al., 2020; Prías-Blanco et al., 2022), and use of a CRISPR RNA-guided integrase system (Vo et al., 2021; Aliu et al., 2022), however, there has been little application of these advanced systems in strawberry (Oosumi et al., 2006).
Some transgene-free methods, such as ribonucleoprotein (RNP) bombardment and RNP transfection of protoplasts, have not been reported in strawberry but have demonstrated success in a range of crops (Liu et al., 2020; Zhang et al., 2021a; Najafi et al., 2023). Other methods of transformation and editing, such as RNA bombardment (Li et al., 2019a) and virus-mediated transformation (Tian et al., 2015) have been sporadically applied in strawberry, indicating a need for further development. Transgene-free targeted mutagenesis approaches for varietal development fall within improved consumer acceptance and reduced regulatory constraints. Thus, continued refinements of genomic tools, transformation and genome editing strategies will position CRISPR/Cas technology as primary tool for gene function validation and crop improvement.
Further development of efficient protoplast isolation, transformation, and regeneration is also important for future transgene-free editing of strawberry. Isolation and regeneration of protoplasts is well established for strawberry (Nyman and Wallin, 1988; 1992a; Barceló et al., 2019; Gou et al., 2020). However, while protocols have been established for transient analyses in protoplasts, few reports of successful regeneration involve transformed materials (Nyman and Wallin, 1992b; Pattanaik et al., 2004; Gou et al., 2020). As such, it is necessary to continue developing methods to transform and regenerate plants from strawberry protoplasts as a foundation for transgene-free genome editing.
As has been established in other species, transformation and regeneration of strawberry depends on several factors, and protocol optimization can be challenging. Experiments to optimize strawberry transformation have observed a wide range of transformation and regeneration efficiencies which vary significantly between genotypes (Zakaria et al., 2014). Other factors, such as transformation method and explant material, have also been reported to impact regeneration efficiency, and response to these factors also varies strongly by genotype (Table 3). Taken together, these findings suggest that some genotype-specific optimization of protocols will be necessary for efficient genetic transformation. Additionally, transformation and regeneration efficiencies tend to be higher for diploid strawberry than octoploid strawberry, even when other factors are held constant between species (Table 3). As Fragaria ×ananassa is the species of economic interest, continued optimization to improve both transformation and regeneration efficiencies will be essential to facilitate genome editing for varietal improvement.
4 Recent advances and resources in CRISPR/Cas-mediated genome editing in strawberryCRISPR, or Clustered Regularly Interspaced Palindromic Repeats, is a genome editing system derived from a bacterial defense network. In bacteria, this defense network operates in two phases to incorporate short fragments of invading DNA into the bacterial genome and then use these sequences to recognize and cleave foreign DNA based on the presence of a protospacer adjacent motif (PAM) (Doudna and Charpentier, 2014; Vigouroux and Bikard, 2020). For genome editing via CRISPR/Cas systems, this bacterial defense pathway is manipulated to target specific sequences within a genome of interest. Unlike other methods of genome editing, such as zinc-finger nucleases (ZFNs) and transcription activator-like nucleases (TALENs), which require substantial protein engineering, CRSIPR/Cas genome editing can be performed simply through a change in the single guide RNA (sgRNA) sequence (Doudna and Charpentier, 2014). The ability to switch editing targets quickly without need for protein engineering has played a major role in the rise of popularity of CRISPR/Cas genome engineering. Double stranded breaks (DSBs) are generated when a Cas endonuclease cleaves DNA at a targeted site using a sgRNA as reference (Cong et al., 2013; Mali et al., 2013; Doudna and Charpentier, 2014). These DSBs can then be repaired through non-homologous end joining (NHEJ), or template mediated homology-directed repair (HDR). NHEJ is error prone, often resulting in insertions or deletions that cause loss of gene function (Cong et al., 2013; Mali et al., 2013; Chen et al., 2022). Targeted mutagenesis using NHEJ mediated repair of CRISPR/Cas-mediated DSBs has been reported in many Rosaceous crops, including apple (Malnoy et al., 2016; Pompili et al., 2020), pear (Charrier et al., 2019; Pang et al., 2019), raspberry (Miller, 2019), and strawberry (Martín-Pizarro et al., 2021; López-Casado et al., 2023). In contrast, template mediated HDR allows precise conversion of targeted single nucleotides or insertion of a specific sequence. Allelic variants differing in single-nucleotide polymorphisms often confer improvement of agronomic traits. HDR pathways can be leveraged to replace alleles with superior variants and has successfully been implemented in crops such as maize, rice, and sugarcane (Shi et al., 2017; Wang et al., 2017; Oz et al., 2021). Reports of HDR-mediated gene targeting are still lacking in strawberry, likely due to low efficiency caused by infrequent occurrence of HDR, competition with NHEJ for DSB repair, and inadequate repair template in close proximity to the DSB site (Chen et al., 2022).
Similar to template mediated HDR, both base and prime editing can generate precision nucleotide substitutions in target genes. Base editing occurs as the result of a catalytically impaired Cas nuclease, such as Cas nickase (nCas) or dead Cas (dCas), fused to a nucleotide deaminase (Molla et al., 2021) and results in an irreversible base conversion without the need for DSBs or an exogenous template (Azameti and Dauda, 2021). Base editing requires the use of different deaminases depending on the desired nucleotide substitution, is currently limited to six of the 12 possible base-swaps, and may result in bystander mutations (Molla et al., 2021). In contrast, prime editing, which occurs as the result of fusing a nCas nuclease with a reverse transcriptase, is capable of generating all 12 possible substitutions as well as small indels in exchange for lower editing efficiency (Molla et al., 2021). Base editing has been applied for the creation of precision nucleotide substitutions in strawberry to support the fine tuning of the sugar content of the strawberry fruit (Xing et al., 2020). While prime editing has not been reported in strawberry, it has been successfully applied in tomato, rice, and wheat (Lin et al., 2020; Xing et al., 2023), demonstrating its potential for precision nucleotide substitution in plant systems.
In addition to generating nucleotide substitutions through base or prime editing, CRISPR/Cas can also be applied to modulate gene expression patterns and epigenetic regulation. By fusing dCas with different effector proteins, it is possible to achieve efficient targeted activation (CRISPRa), repression (CRISPRi), or epigenome modifications (Pan et al., 2021).
While genome editing using the CRISPR/Cas system has largely focused on the use of the Cas9 endonuclease, additional Cas nucleases have been engineered to improve the flexibility of the CRISPR genome editing system by relaxing the requirements for a specific protospacer adjacent motif and altering nuclease function (Guilinger et al., 2014; Shen et al., 2014; Trevino and Zhang, 2014; Tsai et al., 2014; Anders et al., 2016; Komor et al., 2016; 2017; Havlicek et al., 2017; Harrington et al., 2018; Liu et al., 2019; Anzalone et al., 2020; Ghogare et al., 2020; Zhang et al., 2021b; Sukegawa et al., 2023). This is particularly useful for generation of precision nucleotide substitutions using base or prime editing (Wang et al., 2020b; Kantor et al., 2020; Mishra et al., 2020; Huang and Puchta, 2021).
Genome editing is a powerful tool for crop improvement, as it allows for precise, targeted mutation in one or few genes without altering the plant’s genetic background. Genome editing can be an efficient method for varietal improvement as co-editing of multiple genes or alleles by multiplex editing allows researchers to accelerate the generation of desired combinations in elite germplasm without undergoing meiotic recombination (El-Mounadi et al., 2020). In contrast, conventional breeding schemes typically require numerous generations and backcrossing to improve gene or allele combinations for a single trait of interest. Genome editing with CRISPR/Cas also enables the introduction of traits that do not exist within a breeding germplasm. For example, there are currently no widely available breeding sources of resistance to Botrytis cinerea in strawberry, and previous breeding efforts to increase resistance to Botrytis fruit rot (BFR) have been ineffective (Petrasch et al., 2019). However, substantial research has been performed to identify susceptibility genes related to BFR and other strawberry diseases which could make for useful knockout targets in the future.
4.1 Challenges for genome editing in octoploid strawberryWhile the potential benefits of CRISPR/Cas genome editing in Fragaria ×ananassa are numerous, there are several challenges which must be overcome. Octoploid strawberries are highly heterozygous as compared to diploid strawberries (Martín-Pizarro et al., 2019), which can make target gene identification and design of efficient sgRNAs difficult (May et al., 2023). Additionally, the four homoeologous subgenomes of Fragaria ×ananassa are not separated from each other, rather, they have undergone numerous homoeologous exchanges which resulted in increased genomic complexity (Whitaker et al., 2020).
Traditionally, transgene-free genome edited plants are generated through sexual segregation, which is often a labor-intensive and time-consuming process (Gao, 2021). Cultivated strawberries are asexually propagated hybrids, meaning segregation of transgene-free plants through segregation is often impractical. Instead, it is necessary to continue developing other transgene-free genome editing methods for the improvement of strawberry. In addition to optimizing transformation procedures, further development of systems such as the transgene killer CRISPR (TKC), which is able to automatically self-destruct the transgene through inclusion of suicide genes in the CRISPR/Cas construct (He et al., 2018; He et al., 2019; Gu et al., 2021), may enable transgene-free genome editing of strawberry. To date several studies for transgene-free genome editing of Rosaceous crops have been performed (Malnoy et al., 2016; Osakabe et al., 2018; Pompili et al., 2020), though regeneration of explants and selection of transgene-free plants remains a challenge.
4.2 Target traits for CRISPR genome editing in cultivated strawberry (F. ×ananassa)Genome editing in strawberry can be divided into two themes; editing performed in the diploid strawberry and editing performed in the octoploid strawberry. Genome editing is more commonly performed in diploid strawberry due to the simple nature of its genome and its status as a model system for Rosaceae. In the diploid strawberry, genome editing has been successfully employed to manipulate numerous traits (Table 4). Until recently, the complexity of the octoploid genome posed a significant challenge to CRISPR/Cas genome editing, and as such, far less exploration of genome editing in Fragaria ×ananassa has been performed. Due to the availability of new, high-quality octoploid genome assemblies, genome editing has recently been applied for the improvement of several traits (Table 4; Figure 2) and may become a powerful tool for trait discovery and gene characterization in cultivated strawberry.
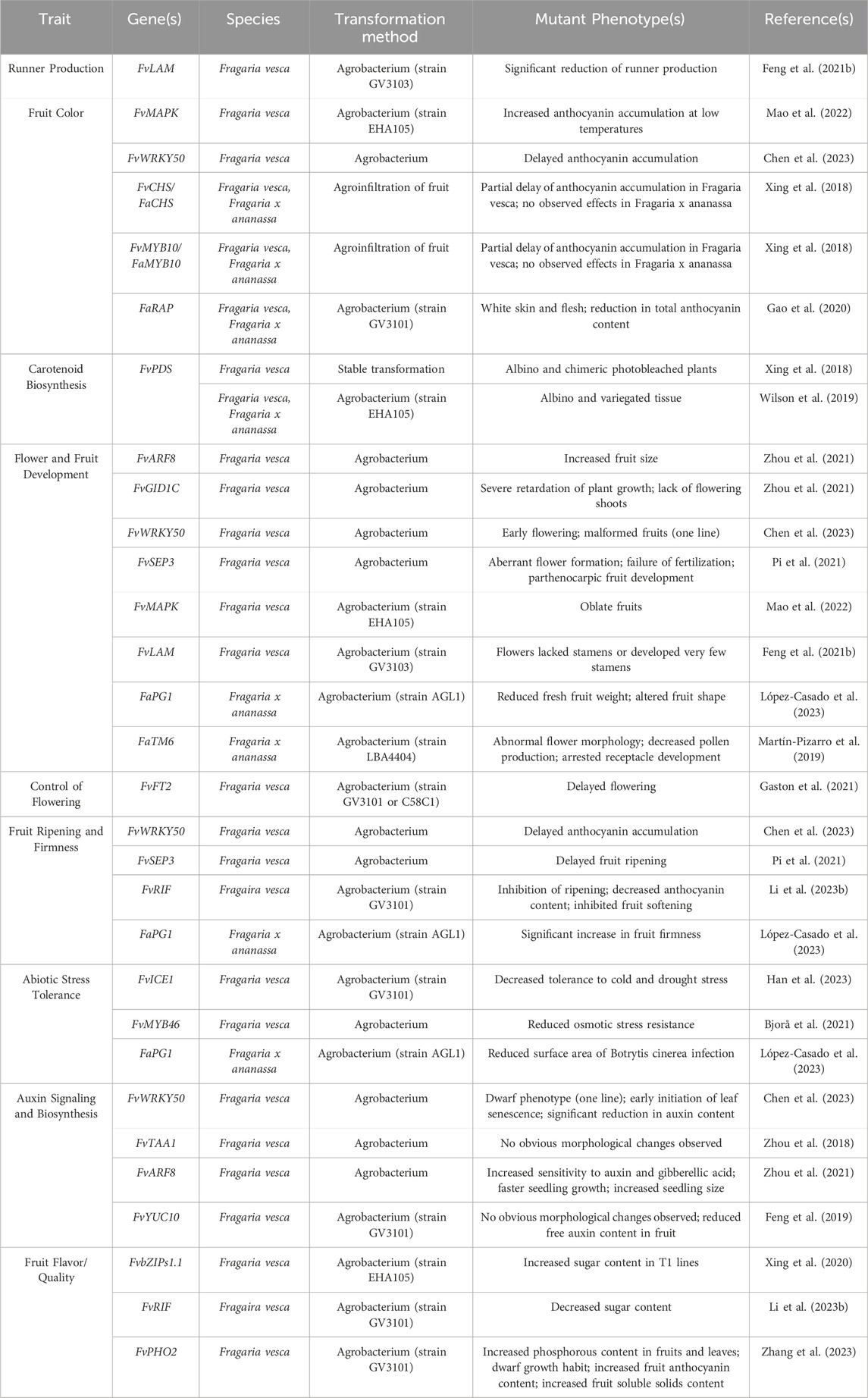
Table 4. Reported CRISPR/Cas-mediated genome editing in strawberry.
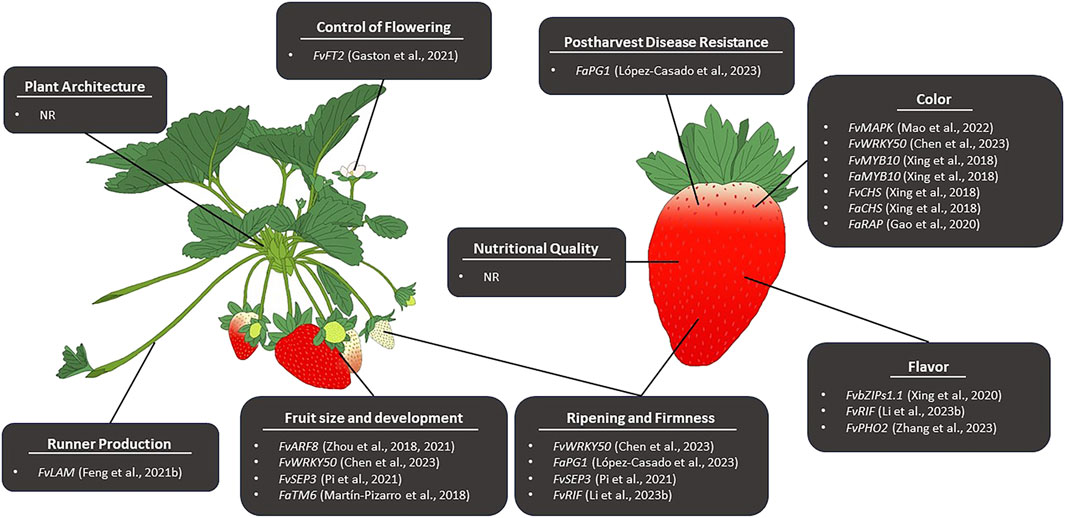
Figure 2. Reported CRISPR/Cas-mediated editing for the improvement of pre- and postharvest traits associated with strawberry fruit quality and production. NR indicates no genome editing has been reported to affect a given trait.
4.2.1 Manipulation of runner production to increase fruit productionIn strawberry, differentiation of runners and branch crowns is mutually exclusive and is influenced by a range of environmental factors (Thompson and Guttridge, 1959; Hytönen et al., 2009; Mouhu et al., 2013; Tenreira et al., 2017; Caruana et al., 2018) However, since each plant produces a limited number of axillary meristems, runner production is considered inversely proportional to fruit production (Tenreira et al., 2017). For this reason, runner removal is a common cultural practice and has demonstrated positive impacts on both fruit quality and yield (Sønsteby et al., 2021). Many studies have attempted to characterize the relationship between fruit and runner production through manipulation of environmental conditions and plant hormones (Hartmann, 1947; Thompson and Guttridge, 1959; Mouhu et al., 2013; Qiu et al., 2019).
As genomic resources became more widely available, studies began to focus on the genes which dictate the decision of flowering versus runnering in strawberry. Suppressor of Overexpression of Constans1 (FvSOC1), FvGA20ox4, FvRGA1, and Loss of Axillary Meristems (FvLAM) have all been identified as runner-associated genes (Mouhu et al., 2013; Tenreira et al., 2017; Caruana et al., 2018; Hytönen and Kurokura, 2020; Feng et al., 2021b). All runner-associated genes which have been identified to date are related to gibberellic acid biosynthesis, suggesting that gibberellic acid plays a major role in the decision between flowering and runnering. The full pathway of gibberellic acid biosynthesis in strawberry remains to be elucidated, however the proposed pathway has undergone continuous expansion as new runnering and flowering-associated genes are identified.
Despite the identification of numerous genes associated with runner production, so far CRISPR/Cas9 genome editing has only been reported in FvLAM in Fragaria vesca (Feng et al., 2021b), and no reports of genome editing of runner-associated genes in Fragaria ×ananassa have been made. CRISPR/Cas9 genome editing has also been applied in tomato and potato for the manipulation of similar traits (Zsögön et al., 2018; Cui et al., 2020; Tang et al., 2022; Tuncel and Qi, 2022), indicating that genome editing can be used effectively to alter plant growth habits. Plant architecture has been shown to have major impacts on yield in numerous crops (Sakamoto and Matsuoka, 2004; Srivastava et al., 2019), making the trait a prime candidate for manipulation via genome editing.
4.2.2 Plant architecture and control of floweringThe timing and duration of flowering play pivotal roles in determining yield potential and harvest season length (Koskela et al., 2012). Manipulation of daylength sensitivity can enable earlier harvest (Soyk et al., 2017) and can even lead to multiple harvests within a single season (de Camacaro et al., 2002; Hancock et al., 2008). During the domestication of strawberry, development of perpetual flowering through photoperiod insensitivity was essential for extending both the range and production period (Gaston et al., 2021). Increased branching can also support increased inflorescences, enabling a greater yield per plant (Premsekhar and Rajashree, 2009). Since the axillary meristem-derived branch crowns are the primary bearer of inflorescences in strawberry, it may be possible to increase fruit yield by increasing floral branching.
Terminal Flower1 (FvTFL1), Flowering Locus T (FvFT2 and FvFT3), and FvWRKY50 were identified as flowering-related genes and shown to interact with each other to control photoperiod response and flowering architecture (Iwata et al., 2012; Koskela et al., 2012; Mouhu et al., 2013; Gaston et al., 2021; Chen et al., 2023). While timing, duration, and development of flowers are important considerations for fruit production, reports of successful editing have been limited to diploid strawberry. FvFT2 and FvWRKY50 have undergone successful genome editing (Gaston et al., 2021), however, editing of other strawberry flowering-related genes has not been reported. Several genes have been identified in other crops with respect to plant architecture and flowering. Branched 1 (AtBRC1), Branched 2 (AtBRC2), SlBRC1b, AtMAX1, AtMAX2, AtCYP79, and AtSPS have been identified to negatively affect branching (Reintanz et al., 2001; Tantikanjana et al., 2001; Stirnberg et al., 2002; Greb et al., 2003; Aguilar-Martínez et al., 2007; Finlayson, 2007; Martín-Trillo et al., 2011). Similarly, Self Pruning 5g (SlSP5G) and Flowering Locus C (BrFLC2) resulted in early flowering and daylight insensitivity (Soyk et al., 2017; Jeong et al., 2019). As no editing of plant architecture or flowering-associated genes has been reported in octoploid strawberry, further functional analysis of known genes is necessary for the improvement of existing varieties.
4.2.3 Fruit developmentFurther study of fruit development genes in strawberry may also enable improvement of fruit production. Parthenocarpy may enable more consistent yields under variable conditions (Hu et al., 2023), and larger fruits tend to be easier to harvest and more desirable to consumers (Hortyński et al., 1991). Both auxin and gibberellic acid have been well established as major regulators of strawberry fruit development (Sharma and Singh, 2009; Liu et al., 2012; Kang et al., 2013; Feng et al., 2019; Fuentes et al., 2019; Zhao et al., 2021) though morphological and environmental factors are also major contributors (Hortyński et al., 1991; Mackenzie et al., 2011; Menzel, 2019; 2021). Through domestication, the average strawberry fruit weight has increased from 1 to 3 g (Fragaria virginiana) to more than 20 g (Fragaria ×ananassa) (Chandler et al., 2012).
Knockout of FvYUC10 altered patterns of auxin accumulation in fruit but resulted in no obvious morphological changes (Feng et al., 2019). RNAi of FvYUC6 was found to negatively affect fruit development (Liu et al., 2014). Other YUC family genes have been identified through transcriptomic studies of strawberry fruit development (Liu et al., 2012; Feng et al., 2019) but have not undergone additional functionalization. Mutations in FvRGA1 and genome editing in Sepallata 3 (FvSEP3) resulted in parthenocarpic fruit development. Genome editing in FvARF8 resulted in increased fruit sizes (Pi et al., 2021; Zhou et al., 2021) and RNAi of FvYUC6 and FveCYP707A4a resulted in reduced fruit sizes (Liu et al., 2014; Liao et al., 2018). Reduction of fruit size was also accomplished through overexpression of Gibberellin Stimulated Transcript (FaGAST1 and FaGAST2) (de la Fuente et al., 2006; Moyano-Cañete et al., 2013). Knockout of Tomato MADS box gene6 (TM6) in both diploid and octoploid strawberry resulted in defects in the anthers and arrested development of the receptacle (Martín-Pizarro et al., 2019), indicating that TM6 also plays a major role in development of both strawberry flowers and fruits.
Significant work has also been performed to explore the mechanisms underlying fruit development in other crops. In apple, a strong QTL for fruit weight was linked to Auxin Response Factor 106 (MdARF106) (Devoghalaere et al., 2012). Silencing of SlIAA7 in tomato resulted in thicker pericarp tissue, and thus, larger fruits (Su et al., 2014). Simultaneous knockout of SlARF8a and SlARF8b resulted in parthenocarpic fruit development and increased parthenocarpic fruit sizes (Hu et al., 2023), and knockout of Fasciated (SlFAS/SlCLV3), Fruit Weight 2.2 (SlFW2.2), and Excessive Number of Floral Organs (SlENO) resulted in increased locule numbers and larger fruits (Zsögön et al., 2018; Yuste-Lisbona et al., 2020). In addition to these genes, some studies have explored the impacts of hormone pathways on fruit development. These studies found that application of gibberellic acid at low concentrations positively impacted speed of development and yield, and decreased production of malformed fruits (Turner, 1963; Sharma and Singh, 2009; Jamal Uddin et al., 2012), further supporting the potential to improve fruit production through manipulation of hormone biosynthesis and signaling.
4.2.4 Fruit flavorStrawberry aroma is the result of a complex mixture of more than 360 volatile compounds (Zabetakis and Holden, 1997; Zorrilla-Fontanesi et al., 2012; Liu et al., 2023), however, only around six odor-active compounds which significantly contribute to flavor have been identified in cultivated strawberry (Raab et al., 2006; Ulrich et al., 2007; Hoffmann et al., 2011). As in many other fruit species, early breeding efforts in strawberry focused on improving firmness and other morphological traits at the expense of flavor and aroma (Hoffmann et al., 2011). As such, new efforts are underway to improve strawberry flavor and aroma.
Several genes associated with strawberry flavor and sugar content have been identified through QTL mapping, genome-wide association studies, and transcriptomic studies (Raab et al., 2006; Chambers et al., 2012; Zorrilla-Fontanesi et al., 2012; Shanmugam et al., 2017; Jiu et al., 2018; Lee et al., 2018; Porter et al., 2023). RNAi of strawberry Chalcone Synthase (FaCHS) paired with overexpression of either Eugenol Synthase (FaEGS) or Isoeugenol Synthase (FaIGS) resulted in partial restoration of wild strawberry aroma (Hoffmann et al., 2011). Additionally, RNAi of Anthranilic Acid Methyl Transferase (FaAAMT), Anthranilate Synthase Alpha Subunit 1 (FaASa1), FaFAD1, and FaTM9 resulted in changes in volatile profiles and soluble solids content (Pillet et al., 2017; Vallarino et al., 2019; Oh et al., 2021; Fan et al., 2022). Overexpression of FaOMT resulted in increased levels of mesifurane, another key volatile compound in strawberry (Fan et al., 2022). Base editing has also been successfully applied to modify fruit sugar content. Xing et al. (2020) used the A3A-PBE base editor to target the conserved sucrose control uORF of FvbZIPs1.1, resulting in 35 novel genotypes that displayed a range of sugar contents.
While flavor is a primary focus of varietal improvement in cultivated strawberry, there are no reports of successful editing of flavor genes to date. However, CRISPR/Cas has been applied in several fruit crops for flavor improvement. Knockout of SlINVIVH1 and SlVPE5 in tomato resulted in increases in sugar content and total soluble solids content in single and double mutant lines (Wang et al., 2021a). Genome editing has also been applied for the improvement of flavor traits in vegetables. Karlson et al. (2022) reported successful application of CRISPR/Cas12a to reduce pungency in Brassica juncea, resulting in increased consumer appeal without reducing nutritional content. This work was performed within the company Pairwise, in Durham, North Carolina, and salad mixes composed of the edited Brassica juncea plants have recently been commercially released (Brown, 2023), indicating commercial potential for genome edited crops with improved flavor traits. These successes highlight the potential applications of genome editing technology for the improvement of flavor in strawberry.
4.2.5 Fruit colorConsumer preferences of fruit color can vary significantly across the globe (Whitaker et al., 2020), and as such, strawberries are available in a wide range of colors. Strawberry coloration is primarily due to variation in accumulation of anthocyanin in the receptacle and achenes during ripening. As fruit color is an important fruit quality trait for consumers, it is a common focus of selection in breeding programs and has undergone substantial investigation to identify associated genes.
Natural mutations in FaMYB10 have been reported as the only natural sources of color variation in strawberry (Castillejo et al., 2020). These findings are supported by those of others, which have identified significant roles of MYB10 and other MYB family genes in controlling anthocyanin accumulation and biosynthesis in strawberry (Whitaker et al., 2020; Denoyes et al., 2023). However, other genes have been reported to impact fruit color in addition to MYB10. Overexpression of FvMYB10 and Reduced Anthocyanins in Petioles (FvRAP) resulted in restoration of anthocyanin biosynthesis in white fruits (Castillejo et al., 2020; Gao et al., 2020). While there is agreement that FvRAP plays a role in regulation of anthocyanin accumulation in addition to FvMYB10, conflicting conclusions have been reached regarding the position of FvRAP in the pathway. Luo et al. (2018) suggested that FvRAP operates downstream of and may be regulated by FvMYB10, whereas Gao et al. (2020) suggested that FvRAP may participate in a color development mechanism separate from FvMYB10. Xing et al. (2018) attempted CRISPR/Cas9 genome editing of FvMYB10 and FvCHS in strawberry through an agroinfiltration of diploid and octoploid fruits but observed no noticeable delay in anthocyanin accumulation in octoploid fruits and only partial delay in diploid fruits. Knockout of FvWRKY50 resulted in downregulation of several anthocyanin-associated genes, including FvMYB10, in addition to delayed anthocyanin accumulation (Chen et al., 2023), and knockout of FvMAPK3 resulted in similar rates of anthocyanin accumulation but higher total anthocyanin content than empty vector controls (Mao et al., 2022).
Genome editing for fruit color modification has also been implemented in other species. In tomato, CRISPR/Cas9-mediated genome editing was used to generate tomatoes that were yellow, pink, and purple in color (Čermák et al., 2015; Filler Hayut et al., 2017; Deng et al., 2018), indicating the potential to fine tune fruit color through the application of genome editing. This may enable a greater range of fruit color options and greater flexibility to cater to consumer preferences around the world.
4.2.6 Nutritional contentStrawberries have a diverse nutritional composition with high levels of biological compounds and phytochemicals (Giampieri et al., 2012; 2013; 2014; Afrin et al., 2016). Strawberries have also been studied for their clinical effects (Afrin et al., 2016). Pigments often add to both nutritive value and antioxidant content (Kapoor et al., 2022). Despite the role of nutritional quality in strawberry popularity, genes underlying nutritional content mechanisms are not widely studied. However, recently, some groups have begun to focus on methods to increase nutritional quality. Integration of wild genotypes into a breeding germplasm has been shown to facilitate improvements in fruit nutritional c
留言 (0)