
記住我
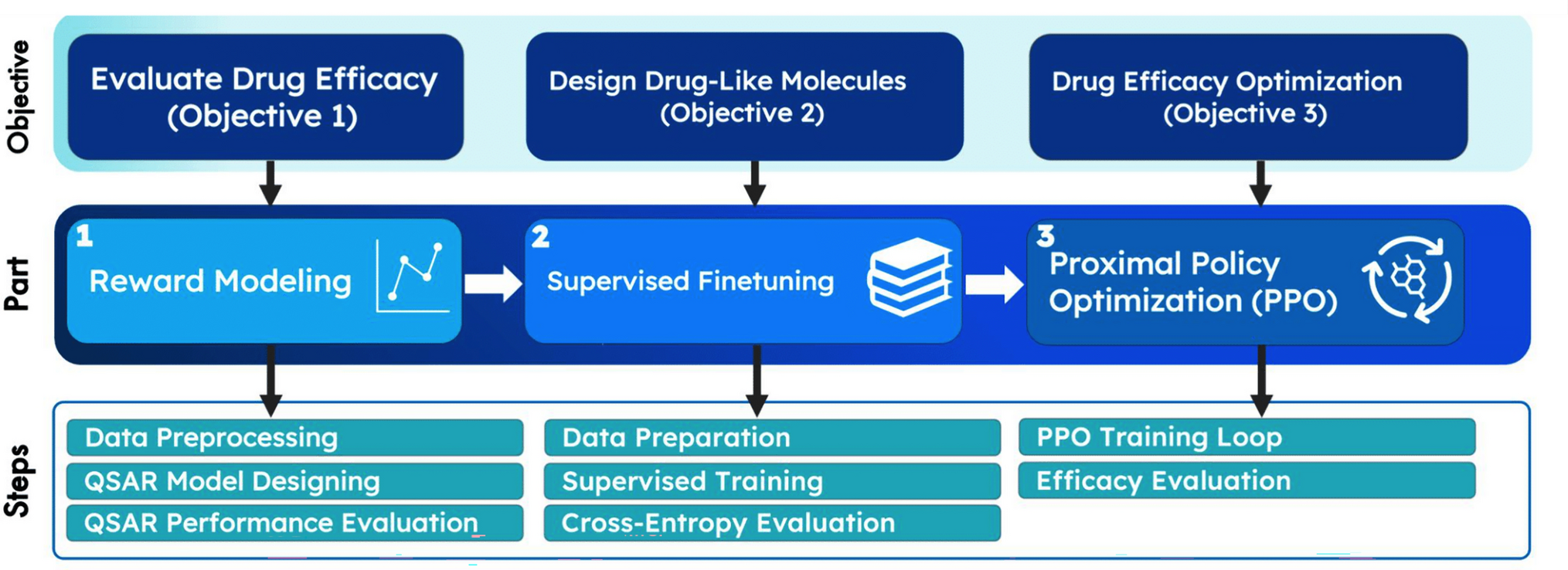





To predict the aggregation propensity of known SCAMs and non-aggregators via molecular dynamics simulation, we constructed a dispersed distribution of 11–12 molecules of a given small organic compound of interest throughout the aqueous solution; we then studied the degree to which spontaneous aggregation occurred over the ensuing trajectory. To calibrate our MD protocol, we studied an initial set of compounds, 1–23 (Fig. 1), for one microsecond in explicit solvent. Over the microsecond trajectory of each system, we identified the number of clusters formed by the solute molecules (Nc).
For a one microsecond simulation of fluconazole 1, the resulting population distribution in regard to the number of clusters formed, Nc, indicates that no clustering was observed, such that all eleven molecules remained separated beyond the 3 Å cutoff that defines a cluster (orange, Fig. 2). This is in agreement with experiment and our previous simulations in water boxes of this [31] and larger size [12]. Conversely, for the microsecond simulation of miconazole 2 in solution, strongly aggregating behaviour was exhibited. The distribution of Nc has a value of around 1, indicating miconazole molecules aggregate to form mainly one large cluster (Fig. 2); again this is in agreement with experiment and previous simulations [12, 31]. These two compounds illustrate the limiting behaviours of a strong aggregator, having a small Nc which approaches 1; and a strong non-aggregator, where Nc approaches the number of solute molecules in the simulation. We note that although these simulations were for 11–12 solute molecules in a box of explicitly modelled water, we have previously performed larger scale simulations of 2 in aqueous solution [12]; these displayed very similar behaviour, such that 99 miconazole molecules initially distributed throughout a larger water box also showed significant levels of aggregation [12].
Fig. 2
Population P (%) of cluster size of molecules 1–23 over 1 µs MD simulation (black) and triplicate MD simulations of 100 ns (orange, red and blue). Compounds that are experimentally observed as non-aggregators were indicated by * next to the molecule label
For the remaining molecules 3–23, a spectrum of aggregation propensity is observed: for example, molecule 18, the insecticide methoxychlor, forms a persistent large aggregate over the microsecond (Fig. 3a) shown by a population profile that is negatively exponential in shape, similar to 2. By contrast, molecule 8, which contains polar piperidine and morpholine rings, shows no propensity to self-associate across the course of the trajectory (Fig. 3b), reflected by a positively exponential Nc population profile, similar to compound 1 (Fig. 2).
Fig. 3
Comparison of MD snapshots of (a) molecule 18 (strong aggregator), (b) molecule 8 (strong non-aggregator), (c) molecule 6 (weak aggregator), and (d) molecule 23 (weak non-aggregator); at time slices of 5 ns, 50 ns, 100 ns and 1 µs of MD simulation. Molecules are distinguished by colour
However, some molecules display intermediate clustering. The population profiles of these molecules do not represent the positive or negative exponential appearance of molecules 1 or 2, but rather a bell-shaped Gaussian-like form, indicating a spectrum of aggregate sizes are populated over the course of the trajectories. Molecule 6 for instance has a tendency to form aggregates, with a dynamic equilibrium between smaller and larger clusters (Fig. 3c). The dynamic nature of these clusters is also evident from the time dependence of Nc (Figure S1) which ranges from around 4–6 clusters initially, to no clusters (at time t = 350 ps) to a completely self-associated system (at t = 630 ps, and again at t = 820 ps). However, the overall tendency of 6 to form fewer larger clusters is clear from the peak in population profile of Nc at around 3 (Fig. 2). Interestingly, experimentally molecule 6, while found to be a SCAM by detergent-based assays, was classified as ambiguous from its scattering behaviour via DLS [1], which may correspond with its simulated intermediate aggregation character. Conversely, molecule 23 exhibits a peak in profile at an Nc value of 8 (Fig. 2) which reflects its propensity towards forming a greater number of small clusters (Fig. 3d).
The cluster population profiles are useful in characterizing the differing degrees of predicted propensity for aggregation of a set of molecules from MD simulation. However, for the purpose of providing a parameter with which to filter molecules as SCAM or non-aggregators, we attempt to classify this behaviour towards forming larger clusters using a suitable metric. To do this, we define fC5 as the fraction of trajectory in which the molecules form fewer than five clusters over the simulation, i.e. Nc <5. Based on fC5, we observe 13 of the 17 experimentally determined aggregators have a fC5 value close to 100% (Fig. 4).
Fig. 4
Comparison of calculated fraction of trajectory forming fewer than five clusters, fC5, for compounds 1–23 over 1 μs (black) with average of three replicas over 100 ns MD simulation in explicit solvent (green) and implicit solvent (purple). The standard deviation bar of calculated fC5 over 100 ns MD simulation, for both explicit and implicit replicas is shown in red. Compounds that are experimentally non-aggregators are signified by the asterisk on top of the related bar
Known aggregators 4, 6 and 11, show more intermediate values of fC5, of 36%, 70% and 50% respectively; this reflects their more polydisperse cluster population profiles (Figs. 2 and 4). All three compounds possess a combination of polar and non-polar functionality (Fig. 5a). Analysis of solute-solute interactions indicates close contacts by atoms across the molecular structures (Figure S2). Interestingly, molecule 11, haematein, has a low logP value of 1.1, lower by 0.6 units compared to similar-sized molecule 17; the latter is a known non-aggregator which shows very limited self-association during MD simulation (Fig. 2). The total polar surface area (TPSA) of 11 is also larger than 17, by 26 Å2 respectively, yet it seems the greater rigidity of 11 promotes transient stacking (Fig. 5b) which is absent in the more conformationally flexible 17.
Fig. 5
(a) Molecular surface for compounds 4, 6, 11 and 17, indicating polar (purple) and nonpolar (green) regions. (b) Representative aggregates from microsecond MD trajectories of 4, 6 and 11
From MD simulations, six molecules have fC5 values of less than 20%: 1, 7, 8, 16, 17 and 23. All six of these molecules are experimental non-aggregators (indicated by the asterisk in Fig. 4). Molecule 16 shows the highest degree of clustering, with a fC5 value of 16% (Fig. 4). In only one case out of the 23 molecule set does MD simulation predict an incorrect outcome: molecule 21 displays strongly aggregating behaviour, with a fC5 value of 92%, whereas experimentally it is classed as a non-aggregator. A replicate microsecond trajectory of 21 exhibited a very similar level of aggregation, with a fC5 value of 89% (Figure S3). The good agreement between microsecond-length replicate trajectories is further illustrated for the weakly aggregating compound 4 and largely non-aggregating molecule 16 (Figure S3).
These microsecond MD simulations successfully predict aggregation propensity for 96% of the molecules in this sample, which includes molecules such as 17 and 23 which are problematic for computational filters, ChemAGG [30] and Aggregator Advisor [26]. However, a protocol using microsecond simulations is somewhat compute-intensive if applied to a larger set of molecules. Therefore, we examined the predictive ability of 100 ns simulations of 1–23 in explicit solvent, performing three replicates. From the cluster population profiles of these shorter simulations (Fig. 2), while in some cases there is a degree of variation between 100 ns replicates, the trajectories capture rather well the aggregation profiles from 1 µs simulation of the data set. This is also reflected by the close agreement in fC5 value across 1–23, as computed from the 1 µs simulation and the 100 ns replicate average (black and green respectively, Fig. 4): the maximum deviation in fC5 estimates is only 10%, found for intermediate aggregator molecule 6.
Besides reducing the length of MD trajectory, to further increase computational throughput, we also explore the effect of using implicit generalized Born solvent model rather than explicitly modelled water molecules. However, while implicit MD simulations predicted qualitatively similar results to the 1 µs or the 100 ns simulations, there were significant quantitative differences for some molecules (Fig. 4). For example, 4 was predicted to be strongly non-aggregating via simulation in implicit solvent, but weakly aggregating from explicit solvent MD, with a fC5 value of 33% from the microsecond trajectory and 31% from the 100 ns replica average (Fig. 4). A trend towards decreased aggregation was also reflected for other molecules: for example, molecules 10 and 11 switched from prediction as aggregators via simulation in explicit water to being misclassified as non-aggregators; molecule 21, the only molecule misassigned by explicit solvent simulations, was (correctly) predicted to be a non-aggregator via implicit solvent. Molecule 6 was an exception, predicted as a strong rather than a weak aggregator in implicit solvent. Overall, it seemed implicit solvent underestimated the propensity for hydrophobic self-association. This was despite using a relatively large surface tension parameter of 0.01 kcal/(mol/Å2) in the solvent model (see Methods).
It appears therefore that MD simulations in explicit solvent are required for identifying SCAMs. In a further effort to reduce computational overhead, rather than use triplicate 100 ns simulations, we consider acquisition of only one 100 ns trajectory per molecule. To evaluate this approach, we extend the test set by a further nine molecules, 24–32 (Fig. 1). For the overall set of 32 molecules, the MD-based assessment using a single 100 ns trajectory yields a success rate of 97%, with only compound 21 being misclassified (Fig. 6). Therefore, all 22 molecules of the previous set are correctly classified by the single 100 ns simulation, as well as all nine new cases: MD simulations correctly identify the aggregators 25, 26, 27 and 30, which have fC5 values of ~ 100% (Fig. 6); and distinguish these SCAMs from the non-aggregators 24, 28, 29, 31 and 32, which have fC5 values of ~ 0%. Therefore, this success rate of 97% for 1–32 compares with a success rate for the same set of 75% via Aggregator Advisor and 72% via ChemAGG (Table S4).
Indeed, the ChemAGG filter misassigns molecule 11 as a non-aggregator. However, as noted earlier, we can observe from MD simulation the subtle propensity of 11 to self-assemble, such that it forms a range of aggregates of two to four molecules in size (Fig. 5b). By contrast, molecules 31 and 32 are known non-aggregators despite having high computed logP values [39], of 4.0 and 3.6 respectively (Table S3). Aggregator Advisor misclassifies 31 and both ChemAGG and Aggregator Advisor fail for 32; the MD protocol correctly identifies both as non-aggregators. Indeed, for these cases, the distribution coefficient logD appears to be a more suitable measure than logP, capturing the lower effective hydrophobicity due to ionization, with logD values of 2.8 and 1.3 respectively (Table S3).
We also note in our previous MD study [31] of 1 and 2 that the higher amphiphilic moment of 2, alongside its greater planarity, reflected the differing aggregation behaviour of these two molecules. We may estimate the amphiphilic moment using the MOE [39] descriptor vsurf_A, which reflects the magnitude of separation between centres of hydrophobic and hydrophilic character in a molecule: miconazole 2 has a vsurf_A value of 6.2 but fluconazole 1 possesses a value of only 3.1. In the set of 32 molecules considered here, there are several known SCAMs with high vsurf_A values, in the range 5–6, namely 2, 12, 15, 16, 19, 22 and 30 (Table S3). These are all correctly predicted as SCAMs by MD. However, Aggregator Advisor misclassifies 16; and ChemAGG misassigns 2, 12 and 16 (Table S4). Interestingly, compounds 8 and 32 also have high vsurf_A values, of 6.7 and 6.3 respectively (Table S3) but are non-aggregators experimentally. The two in silico filters correctly predict 8 as a non-aggregator but misclassify 32; however, MD predicts the behaviour of both 8 and 32 correctly.
Indeed, the vsurf_A value does not seem to predict well aggregator from non-aggregator for 1–32 (Figure S4). Rather, the distribution coefficient logD provides a more discriminative descriptor than vsurf_A or logP, providing rather good agreement with experiment and MD simulation (Figure S4). Indeed, simply assuming aggregation occurs for compounds with a logD > 3 leads to only three compounds in the set being misclassified: compound 11 is misassigned as a non-aggregator, and 21 and 23 as aggregators. Thus, once again, misclassification of 21 is found; we examine this compound further in the following section.
Fig. 6
Comparison of calculated fraction of trajectory forming fewer than five clusters, fC5, over a 100 ns MD simulation in explicit solvent, for compounds 1–32. Experimental non-aggregators indicated by asterisk
Structure-aggregation relationships from MD simulationHere, we apply MD to assess the effect of small changes in chemical structure on predicted aggregation propensity. Firstly, to explore further the misclassification of non-aggregator molecule 21, we conducted a limited in silico structure-property analysis of 21, performing comparative triplicate 100 ns simulations for four closely related derivatives to compute their aggregation propensities. In these simulations, we alter the two phenyl rings of 21 to aliphatic rings (molecule 21d1, Fig. 7a); or replace the two hydroxyl groups on the phenyl rings with methyl groups (21d2). We also change the central part of 21, replacing the tricholoromethyl group with an alkenyl moiety (21d3), or with a hydroxyl function (21d4). Interestingly, the modification of the phenyl rings in 21d1 and 21d2 does not lead to a significant change in the strongly aggregating behaviour of 21 (Fig. 7a).
Fig. 7
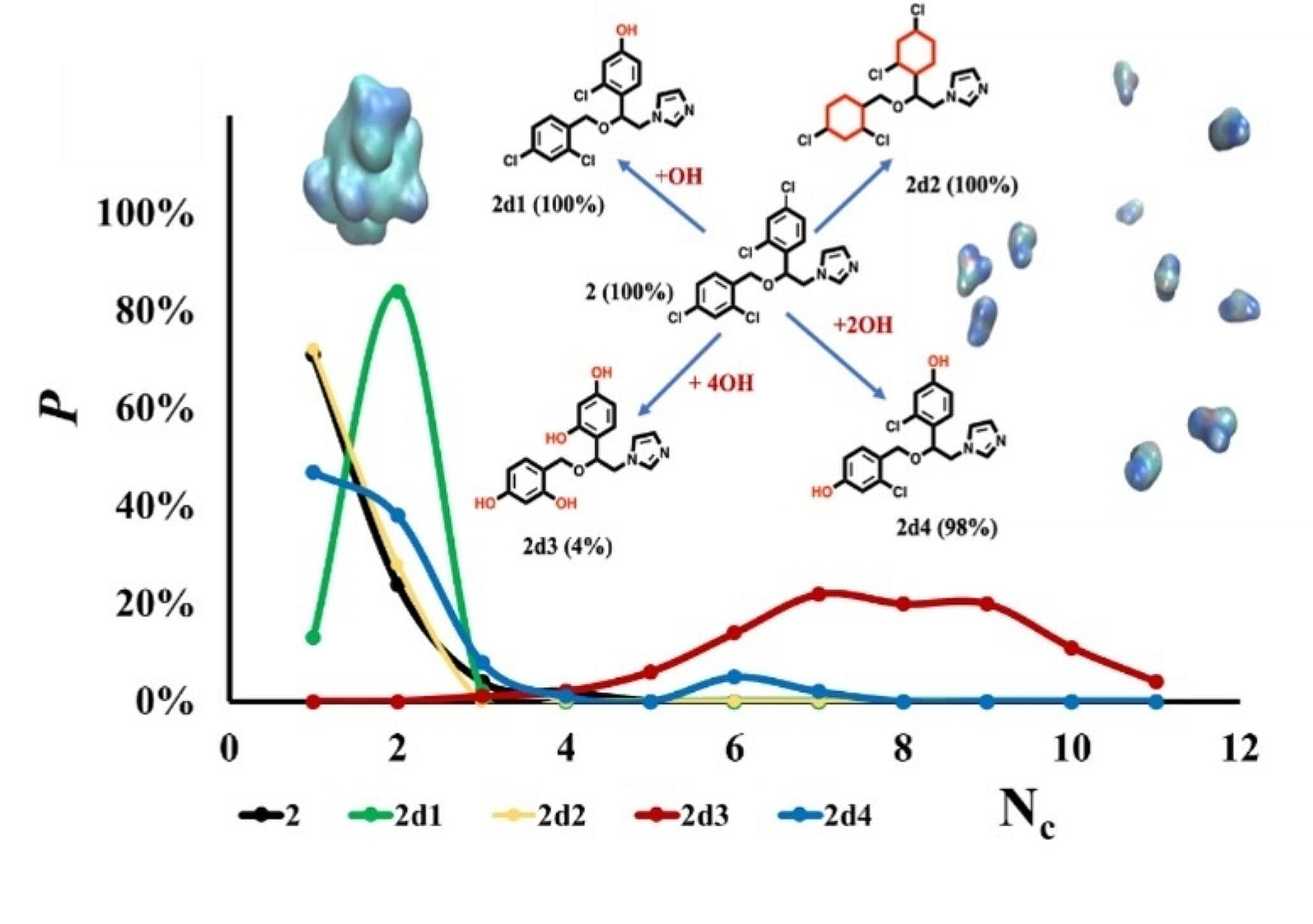
Modification of molecules (a) 21, (b) 1 and (c) 2 and corresponding cluster population P (%) of the compounds and their molecular derivatives as a function of cluster size of 100 ns MD simulations in explicit water. fC5 values are given in parentheses
However, replacement of the trichloromethyl group with the alkenyl moiety in 21d3 reduces aggregation from a fC5 of 100% to a value of 26% (Fig. 7a). Replacement of the trichloro group by a hydroxyl group in 21d4 leads to the elimination of aggregation (fC5 value of 1%). This dramatic switch in simulated behaviour highlights the influence of the largely nonpolar trichloro group on the delicate balance of solute-solute and solute-solvent interactions governing the self-assembly of 21. As observed earlier, changing from explicit to implicit solvent for 21 also leads to a switch to non-aggregating behaviour, again demonstrating the sensitivity of 21 to changes in intermolecular energetics (Fig. 2). We compare 21d2 with the closely related methoxychlor (18): the former only differs from the latter in having two methyl rather than methoxy substituents on the phenyl rings. However, this modest chemical difference is sufficient for MD simulation to predict 21d2 as strongly aggregating and 18, correctly, as strongly non-aggregating. Given the large influence of small changes in chemical structure, the misclassification of 21 by MD may arise from some subtle imbalance in interaction strengths arising from the force field parameters of the solute and solvent.
As a final point, we note that while self-aggregating molecules can be a nuisance in medicinal chemistry hit identification campaigns, such behaviour may be desirable in generating drug delivery systems for example [15]. To illustrate the potentially useful role MD could play in such processes, using the approach taken for 21 above (Fig. 7), we modify the structures of our archetypal strong non-aggregator 1 and aggregator 2: in each case, we seek to reverse their self-association behaviour. Given the nature of this computational experiment, we do not consider the synthetic accessibility of these modifications nor present experimental validation. For the case of 1, removal of the central hydroxyl group did not promote aggregation in MD simulations (1d1, Fig. 7b). However, we find that replacement of the nitrogen atoms by carbon in just one of the two 1,2,4-triazole rings in 1 to give 1d3 was sufficient to entirely reverse its predicted behaviour from non-aggregator (fC5 of 0%) to aggregator (fC5 of 100%). For miconazole 2, replacement of all four chlorine atoms with hydroxyls led to a switch from aggregator (100%) to non-aggregator 2d3 (4%, Fig. 7c). Other substitutions, for example the switch of only one chloro- substituent for a hydroxyl group (2d1) was ineffective in decreasing the self-assembling nature of 2 (Fig. 7c).
留言 (0)