Culture of macrophages and macrophage polarization
Bone marrow derived macrophages were isolated as described previously [24, 25]. The studies were approved by the Animal Ethical Committee of Xi’an Jiaotong University and complied with the Guide for the Care and Use for Laboratory Animals published by the US National Institutes of Health. Six SD rats were sourced from the Experimental Animal Center of Xi’an Jiaotong University (Shaanxi, China) and used for isolation of bone marrow derived macrophages. After the SD rats were killed and soaked in 75% ethanol for 10 min, the femurs and tibias bones were isolated under sterile conditions. Bone marrow was flushed into α-minimum essential medium (αMEM) supplemented with 10% fetal bovine serum (FBS, Gibco, CA, USA) and 1% penicillin-streptomycin (Gibco, CA, USA) by repeated aspiration with 10 ml syringes. The cell suspension was centrifuged at 500 × g for 5 min, and the pellet was resuspended in erythrocyte lysate to lyse red blood cells for 5 min. The cells were washed with αMEM and cultured in αMEM supplemented with 10% FBS, 1% penicillin-streptomycin and 10 ng/ml monocyte colony stimulating factor (M-CSF) for 7 days to allow them to differentiate into M0 macrophages. For M2 polarization, the macrophages were cultured in medium containing 20 ng/mL interleukin (IL)-4 (Sigma-Aldrich, St Louis, MO, USA). After 24 h of induction, the macrophage phenotypes were identified by using flow cytometry (FCM) and quantitative reverse transcription polymerase chain reaction (RT-qPCR) analysis of M2-related markers.
Flow cytometry
The M2 macrophage phenotypes were identified by FCM using antibodies against CD206. Briefly, the cells were incubated with an anti-CD206 antibody (Invitrogen) for 30 min at 4 °C. For immunolabeling, the cells were then incubated with a goat anti-rabbit IgG (H + L) cross-absorbed secondary antibody, FITC (Invitrogen), for 30 min at 4 °C in the dark. Excess antibody was removed by washing the cells with PBS. Then, the cells were analysed by flow cytometry on a FACSCalibur system.
Isolation and identification of exosomes
Exosomes were purified from M2 macrophages by differential ultracentrifugation as previously described [26]. Briefly, the conditioned medium was centrifuged at 300 × g for 10 min and 16,500 × g for 20 min to remove debris and dead cells, followed by filtration through a 0.22 μm filter. Then, the exosomes were pelleted by ultracentrifugation at 120,000 × g for 70 min at 4 °C, resuspensed in PBS and stored at -80 °C for subsequent experiments. The morphology (cup-shaped) of the exosomes was examined using transmission electron microscopy(TEM). The size distribution of the exosomes was verified using a NanoSight NS300 (Marvel, UK). To determine the typical surface markers of exosomes, including CD9 and TSG101, Western blotting was performed.
Isolation and culture of bone marrow-derived mesenchymal stem cells
Rat bone marrow-derived mesenchymal stem cells were isolated as described previously [26]. Three SD rats were used in this study. Briefly, the femurs and tibias were resected under sterile conditions. Bone marrow was flushed out by repeated aspiration with syringes. After centrifugation at 800 rpm for 5 min, the collected cells were seeded into 6-well dishes and cultured in αMEM supplemented with 10% fetal bovine serum (FBS, Gibco, CA, USA) and 1% penicillin-streptomycin (Gibco, CA, USA) in an incubator with 5% CO2 at 37 °C. Cells at passages 3–5 were used in subsequent experiments.
Cell proliferation assay
Cell Counting Kit-8 (CCK-8) assays (Dojindo, Kumamoto, Japan) were used to assess the effects of M2-Exos on the proliferation of BMMSCs. BMMSCs were seeded into 96-well plates at a density of 2 × 103 cells/well and treated with culture medium containing M2-Exos (50 µg/mL) or PBS at the same volume for 2 days. Ten microlitres of CCK-8 solution was added to each well. After incubation for another 3 h, the absorbance was measured at 450 nm with a spectrophotometer.
Alkaline phosphatase (ALP) activity assay, ALP staining and Alizarin Red staining(ARS)
To assess the effects of M2-Exos on osteogenic differentiation of BMMSCs, ALP activity assays, ALP staining and Alizarin Red staining were performed according to a previously reported protocol [27]. BMMSCs were cultured in osteogenic medium (α-MEM containing 10% exosome-free FBS, 10 nM dexamethasone, 50 µM L-ascorbic acid-2-phosphate, and 10 mM β-glycerophosphate; all from Sigma) supplemented with M2-Exos (50 µg/mL), or the same volume of PBS. After 7 days, the ALP activity was determined using p-nitrophenyl phosphate (p-NPP, Sigma) as a substrate and quantified colorimetrically at 405 nm. After 7 days of osteogenic induction, ALP staining was performed to analyse the osteoblastic differentiation. After 14 days, alizarin red staining was performed. For the quantitative assessment of alizarin red staining, the absorbance at 590 nm was detected using a spectrophotometer.
Quantitative reverse transcription polymerase chain reaction analysis (RT-qPCR)
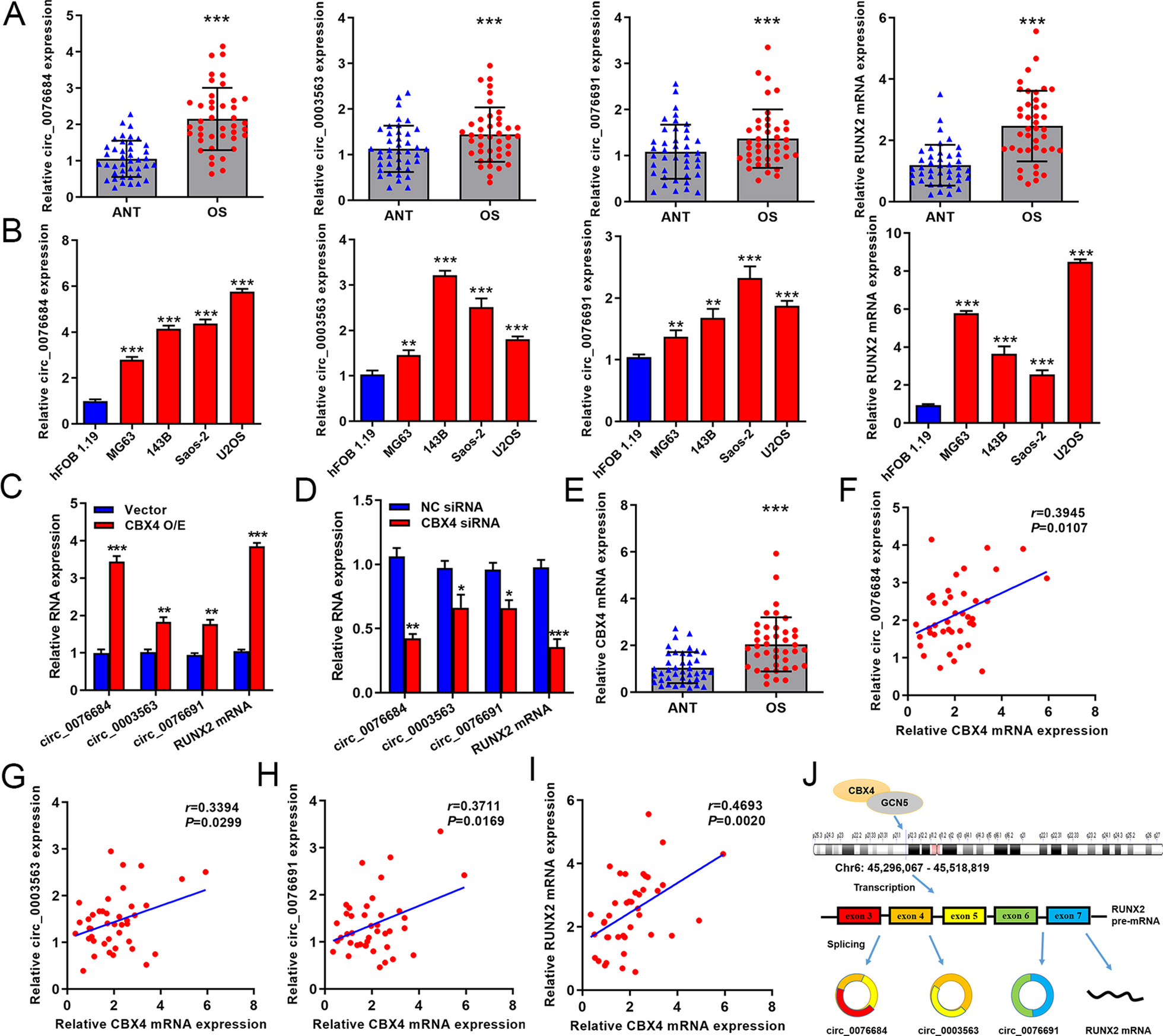
Total RNA was extracted from cells using TRIzol reagent (Invitrogen). cDNA was synthesized by using a PrimeScript RT kit (Takara Bio, Dalian, China). RT-qPCR was performed with an ABI 7500 system (Applied Biosystems). GAPDH mRNA levels were used for normalization. Relative gene expression levels were calculated by the comparative 2−ΔΔCt method. The primers used were as follows: 5ʹ-GCCAGCCTCGTCTCATAGACA-3ʹ (forwards), 5ʹ-AGAGAAGGCAGCCCTGGTAAC-3ʹ (reverse) for GAPDH; 5ʹ-CGAATAACAGCACGCTATTAA-3ʹ (forwards), 5ʹ-GTCGCCAAACAGATTCATCCA-3ʹ (reverse) for RUNX2; 5ʹ-ACCCTCTCTCTGCTCACTCTGCT-3ʹ (forwards), 5ʹ-GCTCCAACTCCATTGTTGAGGTAG-3ʹ (reverse) for OCN; 5ʹ-AGGGTTACTTGGGTTGCC-3ʹ (forwards), 5ʹ-GGGTCTTCAGCTTCTCTCC-3ʹ (reverse) for IL-10; 5ʹ-CAGTATTCACCCCGGCTA-3ʹ (forwards), 5ʹ-CCTCTGGTGTCTTCCCAA-3ʹ (reverse) for Arg-1; 5ʹ-TGTTTTGGCTGGGACTGACCTA-3ʹ (forwards), 5ʹ-CGGGTGTAGGCTCGGGTAGTAG-3ʹ (reverse) for CD206.
Establishment of a rat model of steroid-induced ONFH
Sixty healthy male SD rats (age: 8–10 weeks; body weight: 260–320 g) were randomly divided into three groups (n = 20 per group): the Normal group, Model group and M2-Exos group. The rat steroid-induced ONFH model in an early stage was established by using lipopolysaccharide combined with methylprednisolone according to published protocol [28]. Rats in Model group and M2-Exos group were intravenously injected twice with 4 mg/kg lipopolysaccharide (LPS, Sigma, St. Louis, MO, USA) at a time interval of 24 h. Twenty-four hours later, the animals were intramuscularly injected with 60 mg/kg methylprednisolone (MPS, Pfizer, New York, USA) once a day for 3 days. Rats in Normal group were treated with the same volume of saline buffer as those in Model group. Rats in M2-Exos group were intravenously injected with 100 µg of exosomes derived from M2 macrophages every week for 4 weeks. The dosage and timing of M2-Exos was based on references from previous literature [26, 29, 30]. At the same time, rats in Model group were intravenously injected with an equal volume of PBS. Four weeks after the last injection of MPS, the rats were randomly selected for further study.
Hematological examination
Four weeks after the last injection of MPS, blood sample was obtained. The levels of inflammatory markers including TNF-α, IL-6 and IL-10 were quantified by commercially available ELISA kits (R&D Systems, USA)according to the manufacturer’s instructions. The optical density was measured at 450 nm using a spectrophotometer (Thermo). Then, concentrations, expressed as pg/mL, were calculated according to the optical density and the standard curve.
Hematoxylin-eosin (HE) staining
Four weeks after the last injection of MPS, ten rats of each group were euthanized and the femoral heads were collected from both sides. The left samples were fixed in 10% neutral buffered formalin for one week, decalcified with 10% EDTA for four weeks, dehydrated through graded ethanol solutions, embedded in paraffin, cut into 4-µm sections and stained with hematoxylin-eosin. The osteonecrotic changes in every group were observed by two experienced observers using a light microscope in a blinded fashion. The evaluation criteria for osteonecrosis in this model was based on previous reports [31, 32]. Osteonecrosis was judged according to the presence of empty lacunae or pyknotic nuclei of osteocytes within the trabecular bone, along with the accumulation of hypertrophy fat cells and debris of bone marrow cells. The rate of empty lacunae was calculated in five randomly selected fields per section. The right samples were stored for Micro-CT Scanning.
Immunohistochemical staining and TRAP staining
To evaluate osteogenic and adipogenic activity, immunohistochemical staining for osteocalcin(OCN) and peroxisome proliferator-activated receptor γ (PPAR-γ) was conducted. In addition, to evaluate angiogenesis, immunohistochemical staining for vascular endothelial growth factor(VEGF) was performed. Furthermore, to assess inflammatory reactions, immunohistochemical staining for TNF-α, IL-6 and IL-10 was performed. Images were quantitatively analyzed with Image-Pro Plus Software. The mean density was calculated(defined as the ratio of integrated optical density to total area).
To assess osteoclast activity, tartrate-resistant acid phosphatase (TRAP) staining was conducted using a TRAP staining kit from Sigma-Aldrich. TRAP-positive cells were labelled with purplish red. Five sections were taken from each rat and ten optical fields (magnification: ×100) in each specimen were randomly selected for the evaluation of number of osteoclasts. Active osteoclasts were defined as TRAP-positive multinuclear cells containing three or more nuclei.
Double fluorescence labelling of tetracycline and calcein
Five rats were injected subcutaneously with tetracycline hydrochloride (25 mg/kg, Sigma-Aldrich, St. Louis, MO, USA) to mark sites of bone formation on the 14th and 13th days before sacrifice. Ten days later, the rats were injected subcutaneously with the second bone marker calcein (10 mg/kg, Sigma-Aldrich, St. Louis, MO, USA) on the 4th and 3rd days before sacrifice. Then, the samples were collected, fixed in 10% formalin solution, embedded in methyl methacrylate and polished. The sections were visualized and photographed under a fluorescence microscope. The mineral apposition rate (MAR, µm/d) was calculated by dividing the tetracycline (yellow) and calcein (green) double-labelling interval by the administration time interval(10 days).
Micro-CT scanning
Four weeks after the final injection of MPS, the femoral heads were dissected from the rats and scanned by high-resolution micro-CT (eXplore Locus SP, GE, USA) with a resolution of 14 μm per pixel. Three-dimensional (3D) reconstruction was accomplished using a Reconstruction Utility. The region of interest (ROI) was selected in the femoral head without the cortical shell and quantitative parameters including bone mineral density (BMD), bone volume/total volume of bone (BV/TV), trabecular number (Tb.N), and trabecular separation (Tb.Sp) were calculated to evaluate the microarchitecture of the trabecular bone in the femoral heads.
Angiography
Micro-CT-based angiography was performed in five rats to detect the blood supply of the femoral head as previously reported [26]. After anesthesia, the abdominal aorta and vein were dissected. Microfil MV-122(Flow Tech, Carver, MA, USA) was perfused into the distal abdominal aorta to visualize vessels within the femoral head. After being placed at 4 °C for 24 h, the bilateral femoral heads were obtained and decalcified in 10% EDTA for four weeks until the femoral heads could be easily pierced with a pin. Then, the samples were scanned by micro-CT as described above. Three-dimensional reconstruction of blood vessels was accomplished, and the vascular volume was calculated.
Immunofluorescence staining
To study M1 and M2 macrophage density, sections were analysed for the specific marker of the M1 phenotype (CD86) and the marker of the M2 phenotype (CD206) by immunofluorescence staining. Briefly, the sections were incubated with anti-CD86 (1:200; Abcam) and anti-CD206 (1:1000; Abcam) primary antibodies at 4 °C overnight, followed by staining with a fluorescein-conjugated antibody (1:200, DAKO) for 1 h at room temperature. Then, the sections were treated with 4,6-diamino-2-phenylindole (DAPI) to stain the nuclei. All images were obtained using a fluorescence microscope(Olympus). Quantitative analysis of CD86-positive macrophages (M1) and CD206-positive macrophages (M2) in each group was performed by two researchers who were blind to the groups. The number of M1 and M2 phenotype macrophages was counted at 200× magnification by selecting ten random fields per slide. Based on five independent replicates of each group, the percentages of M1 and M2 phenotype macrophages were calculated as the percentages of CD86-positive cells/total DAPI-positive cells and CD206-positive cells/total DAPI-positive cells, respectively.
Statistical analysis
All data were analysed by SPSS 18.0. Categorical data were analysed using Pearson’s chi-square test. All the numerical data are shown as the mean ± standard deviation (SD). Student’s t test was used to compare numerical data between two groups, and one-way analysis of variance (ANOVA) was used to compare numerical data among multiple groups. p < 0.05 was considered statistically significant.





留言 (0)