Pulmonary hypertension (PH) is a chronic life-threatening disorder, characterized by excessive pulmonary arterial remodeling and progressive increases in pulmonary arterial pressure, leading to eventual right heart failure1. Pulmonary vascular remodeling in PH involves complex mechanisms, including dysfunction of pulmonary arterial endothelial cells (PAECs), altered crosstalk between vascular cells, hypoxia, sustained inflammation, and abnormal proliferation of pulmonary arterial smooth muscle cells (PASMCs)2. Although various drugs targeting nitric oxide, endothelin-1 and prostacyclin pathways have been used to improve pulmonary circulation, these drugs failed to reverse pulmonary arterial remodeling or decrease overall PH mortality3. Therefore, novel endogenous pathogenic factors and therapies for pulmonary vascular remodeling and PH are urgently needed.
Increasing evidence has revealed pulmonary endothelial dysfunction as a central factor in pulmonary vascular remodeling and PH. Various types of programmed cell death, including apoptosis, autophagy, necroptosis and pyroptosis, have been reported to be involved in PH development4, 5, 6. In particular, PAEC apoptosis drives the initiation and progression of pulmonary arterial hypertension, which leads to the loss of distal arteriolar integrity or the selection of hyperproliferative, apoptosis-resistant endothelial cells that may contribute to "angioproliferative" plexiform lesions7, 8. Genetic evidence has shown that loss-of-function mutations in BMPR2 activate PAEC apoptosis and contribute to spontaneous generation of PH in mice by eliminating the antiapoptotic effects of BMP99. Moreover, the two most common animal models of PH, monocrotaline (MCT)- and chronic hypoxia-SU5416 (HySu)-induced rat models, are initiated by endothelial cell apoptosis and dysfunction10, 11. Thus, therapeutic strategies aimed at endothelial repair and repression of PAEC apoptosis may be effective in PH treatment. However, the endogenous pathogenic factor that activates PAEC apoptosis and PH progression remains poorly understood.
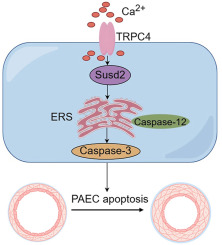
Canonical transient receptor potential (TRPC) channels, a family of Ca2+-permeable channels, play an important role in various diseases. The seven mammalian TRPC members TRPC1-7 are globally expressed and participate in a broad spectrum of cellular functions and physiological processes. As store-operated Ca2+ entry modulators, some TRPC channels have been reported to be involved in PH by contributing to pathological PASMC phenotypes or endothelial hyperpermeability12, 13, 14. To date, the regulatory role of TRPC channels in endothelial cell survival is not yet elucidated. In this study, we revealed the elevation of TRPC4 rather than other TRPC members in PAECs from HySu-induced PH mice and MCT-treated PH rats, as well as hypoxia-exposed PAECs. TRPC4 activated PAEC apoptosis in a caspase-12/endoplasmic reticulum stress (ERS)-dependent manner by promoting the expression of the proapoptotic protein sushi domain containing 2 (Susd2). Inhibiting TRPC4 ameliorated PAEC apoptosis and PH progression in animals by repressing Susd2 signaling, which may serve as a therapeutic target for the management of pulmonary vascular remodeling and PH development.



留言 (0)