
記住我




The Fusarium oxysporum species complex (FOSC), ranked fifth among the top 10 plant pathogens (Dean et al., 2012), causes devastating vascular wilt diseases in over 150 economically important crops, including tomato, cotton, banana, cucumber, melon, and watermelon, leading to significant global yield losses (Di Pietro et al., 2003; Michielse and Rep, 2009; Gordon, 2017; Husaini et al., 2018). FOSC comprises over 100 formae speciales (f. sp.), each displaying distinct pathogenicity toward various host plant species (Gordon, 2017). These strains can be categorized as either plant pathogenic or nonpathogenic, displaying morphological variations (Edel-Hermann and Lecomte, 2019). FOSC strains exhibit strong host specificity toward a limited number of plant species, although some strains from different evolutionary lineages can infect the same host plants (Ma et al., 2013). During the infection process, soil-inhabiting FOSC fungi adhere to the plant root surface, undergo hyphal growth, penetrate the root epidermis, and eventually colonize the xylem vessels (Di Pietro et al., 2003; Michielse and Rep, 2009) This colonization disrupts various physiological, biochemical, and metabolic events, leading to wilting and eventual death of the invaded plants.
In the past two decades, extensive research has focused on understanding the molecular mechanisms underlying pathogenicity of F. oxysporum during its interactions with tomato or Arabidopsis. Throughout the infection process, F. oxysporum induces alterations in extracellular and intracellular pH (Masachis et al., 2016; Fernandes et al., 2023), stimulates the production of reactive oxygen species (ROS) by the NADPH oxidase B complex to facilitate chemotropic growth (Nordzieke et al., 2019), and neutralizes plant-derived ROS (Zhang et al., 2023). Simultaneously, F. oxysporum secretes small effector proteins into the xylem, collectively known as Secreted in Xylem (SIX), to disrupt and circumvent plant immunity, thereby enhancing the fungal pathogenicity (de Sain and Rep, 2015; Jangir et al., 2021). Researchers have characterized 14 SIX genes in F. oxysporum f. sp. lycopersici (Houterman et al., 2007; de Sain and Rep, 2015). Among these, FolSIX1, FolSIX3, FolSIX5, FolSIX6, and FolSIX8 are crucial for full pathogenicity of F. oxysporum f. sp. lycopersici in susceptible tomato plants (Jangir et al., 2021). Furthermore, extensive studies have been conducted on various pathogenicity-related components, including transcription factors, governing pathogenicity functions in F. oxysporum (Husaini et al., 2018; Zuriegat et al., 2021). For instance, FoCon7-1, FoSge1, FoFtf1, Folctf1, Folctf2, and FolCzf1 play critical roles in F. oxysporum pathogenicity, likely by regulating the expression of effector genes (Michielse et al., 2009; Bravo-Ruiz et al., 2013; Ruiz-Roldán et al., 2015; Yun et al., 2019). Despite these significant advances, a comprehensive understanding of the molecular mechanisms and regulatory network governing F. oxysporum pathogenicity on distinct host plants remains to be fully established.
Post-translational modifications (PTMs), including phosphorylation, ubiquitination, glycosylation, acetylation, and lipidation, are pivotal for regulating the development, stress responses, and pathogenicity of plant pathogenic fungi. These PTMs profoundly influence the biochemical functions of proteins, affecting their activity, stability, and subcellular localization (Liu et al., 2021). For instance, N-glycosylation, catalyzed by N-glycosyltransferase Gnt2, is indispensable for the morphogenesis and virulence of F. oxysporum (Lopez-Fernandez et al., 2015). Additionally, palmitoyl transferase FonPAT2-catalyzed palmitoylation of the FonAP-2 complex influences the stability and interactions among core subunits of the complex, playing a vital role in growth, development, stress responses, and pathogenicity of F. oxysporum (Xiong et al., 2023). Lysine acetyltransferase FolArd1-mediated acetylation regulates the stability of the effector FolSvp1, thereby implicating its role in the pathogenicity of F. oxysporum (Li et al., 2022). Further exploration and characterization of various PTMs on pathogenicity-related proteins will undoubtedly yield new insights, shedding light on the molecular mechanisms underpinning F. oxysporum pathogenicity.
Poly(ADP-ribosyl)ation (PARylation), a PTM process, was initially discovered in the 1960s (Chambon et al., 1966; Hasegawa et al., 1967) and has since been found ubiquitous in various organisms (Alemasova and Lavrik, 2019). PARylation primarily relies on poly(ADP-ribose) polymerases (PARPs), which transfer ADP-ribose moieties from nicotinamide adenine dinucleotide (NAD+) to target proteins or themselves, resulting in the formation of linear or branched poly(ADP-ribose) polymers on glutamate (Glu), aspartate (Asp) or lysine (Lys) residues (D’Amours et al., 1999; Gibson and Kraus, 2012; Bai, 2015). Notably, lysine at 988 position (E988) of PARP1 is an important site that is associated with the occurrence of self-PARylation and mutation of this lysine led to a failure in creating PAR chain elongation, only adding a single mono-ADP-ribosyl (MAR) to the site (Chen et al., 2019). Conversely, these covalently attached polymers can be enzymatically degraded by poly(ADP-ribose) glycohydrolase (PARG) through endo- and exo-glycosidase reactions (Meyer-Ficca et al., 2004). PARylation has been confirmed to play essential roles in numerous cellular processes, including DNA repair, transcription regulation, chromatin modification, and ribosome biogenesis (Gibson and Kraus, 2012; Bock et al., 2015; Dasovich and Leung, 2023). Moreover, protein PARylation has implications in plant immunity against different pathogens (Feng et al., 2015; Song et al., 2015). For example, disrupting AtPARPs or AtPARGs modifies Arabidopsis responses to biotic and abiotic stresses (Adams-Phillips et al., 2010; Feng et al., 2015; Song et al., 2015, Yao et al., 2021). However, in filamentous fungi, PrpA, a putative PARP homolog in Aspergillus nidulans, functions early in the DNA damage response (Semighini et al., 2006). It was previously shown that knocking out FolPARG1 had no discernible effect on the pathogenicity of F. oxysporum f. sp. lycopersici (Araiza-Cervantes et al., 2018). To date, the biological functions of PARPs and protein PARylation in plant pathogenic fungi remain largely unexplored.
Kin4, a protein kinase, functions as a mother cell-specific SPOC (spindle position checkpoint) component. It plays crucial role in regulating the MEN (mitotic exit network) activity and contributes to delaying cell cycle progression with spindle misalignment (D’Aquino et al., 2005; Pereira and Schiebel, 2005). In the yeast Saccharomyces cerevisiae, the deletion of ScKin4 reduces the survival of Δkar9 cells and exhibits a modest impact on mitotic progression under normal growth conditions (Tong et al., 2004; D’Aquino et al., 2005). In the filamentous fungus Aspergillus nidulans, KfsA, a homolog of ScKin4, proves essential for proper asexual spore formation (Takeshita et al., 2007). As Kin4 is a protein kinase, it is noteworthy that ScKin4 possesses the ability to phosphorylate with a catalytic site T209 located in the active loop of ScKin4 (D’Aquino et al., 2005; Caydasi et al., 2014). However, the biological functions of Kin4 and its biochemical relationship with PARP1 in plant pathogenic fungi remain poorly understood.
Fusarium oxysporum f. sp. niveum (Fon) causes devastating vascular Fusarium wilt disease, posing a significant threat to the global watermelon industry (Martyn, 2014; Everts and Himmelstein, 2015). In this study, our objective was to explore the functions of PARylation-related enzymes, FonPARP1 and FonPARG1, in Fon pathogenicity and elucidate their interplay with FonKin4. Our results demonstrate that both FonPARP1 and FonKin4 play critical roles in Fon pathogenicity, with FonKin4-mediated phosphorylation of FonPARP1 being a key regulatory mechanism. These findings underscore the significance of protein PARylation, facilitated by the FonKin4-FonPARP1 cascade, within the regulatory network governing pathogenicity in Fon and other plant pathogenic fungi.
2 Materials and methods 2.1 Fungal strains and growth conditionsThe wild-type (WT) strain, Fon race 1 strain ZJ1, was used for generating deletion mutants (Gao et al., 2022). Experiments related to fundamental biological processes and stress responses were conducted as previously reported (Dai et al., 2016). For assessing growth, Fon strains were cultivated on potato dextrose agar medium (PDA) or minimal medium (MM) at 26°C for 7 d (Gao et al., 2022). Conidiation and germination assays were performed using mung bean liquid (MBL) and yeast extract peptone dextrose (YEPD) at 26°C for 2 d and 12 h, respectively (Gao et al., 2022). To observe conidial morphology and septation, macroconidia were stained with 10 μg/mL calcofluor white (CFW) and observed under a Zeiss LSM780 confocal microscope (Gottingen, Niedersachsen, Germany) (Gao et al., 2022). For stress response assays, Fon strains were cultivated on PDA supplemented with 0.7 M sodium chloride (NaCl; Sinopharm Chemical, Shanghai, China), 0.7 M calcium chloride (CaCl2; Sinopharm Chemical), 3 mM paraquat (Syngenta Crop Protection, Basel, Switzerland), 5 mM hydrogen peroxide (H2O2; Sigma-Aldrich, St. Louis, MO, United States), 0.2 g/L Congo red (CR; Sigma-Aldrich), 0.2 g/L Calcofluor white (CFW; Sigma-Aldrich) or 0.3 g/L sodium dodecyl sulfate (SDS; Sigma-Aldrich) for 7 d. The mycelial growth inhibition rate (MGIR) was calculated as described previously (Tang et al., 2018).
2.2 Generation of targeted deletion mutants and complementation strainsDeletion and complementation vectors were constructed as previously described (Gao et al., 2022). To generate targeted deletion mutants, the upstream and downstream flanking sequences of the target genes were amplified and then fused with the HPH (Hygromycin B phosphotransferase) fragment through a double-joint polymerase chain reaction (PCR). The PCR products were subsequently transformed into WT protoplasts. Transformants were selected on PDA containing 100 μg/mL hygromycin B (Roche, Basel, Switzerland) and further verified through PCR and Southern blotting. To generate complementation vectors, genomic fragments containing ~1.5 Kb native promoter and open reading frame (without a stop codon) of FonPARP1, FonKin4, and their mutated variants were co-transformed with XhoI-digested pYF11-neo plasmid into the yeast strain XK-125 using the Alkali-Cation Yeast Transformation Kit (MP Biomedicals, Solon, OH, United States). The resulting recombinant vectors were introduced into protoplasts of the corresponding deletion mutants. Transformants were selected on PDA supplemented with 50 μg/mL neomycin (Sangon Biotech, Shanghai, China) and confirmed by PCR amplification or Western blotting with anti-GFP antibody (Cat. #ab290, Abcam, Cambridge, United Kingdom) or anti-GAPDH antibody (Cat. #EM1101, HuaBio, Hangzhou, China). Site-specific point mutated variants FonPARP1E729K and FonKin4T462A were created using Mut Express MultiS Fast Mutagenesis Kit (Vazyme Biotech, Nanjing, China).
2.3 Disease assays and fungal biomass estimationDisease assays were performed using a previously established protocol (Dai et al., 2016). Watermelon (Citrullus lanatus L. cv. Zaojia) plants were grown in a potting mix (vermiculite: plant ash: perlite = 6:2:1) in a growth room with a 16-h light/8-h dark photoperiod. For inoculation, three-week-old plant roots were dipped in spore suspensions (5 × 106 spores/mL) of different Fon strains for 15 min, subsequently replanted in soil, and covered with plastic wrap for 3 d. Disease symptoms and progress were assessed using a 4-scale rating standard (0 = no symptom, 1 = chlorosis, 2 = wilting, 3 = death). For tissue examination, 1 cm segments of roots and stems were collected from the inoculated plants at 15 d post-inoculation, sterilized by immersion in 70% ethanol for 30 s, and then placed on PDA for incubation at 26°C for 3 d (Gao et al., 2023). Colony morphology, mycelial color and conidia characteristics were carefully examined to distinguish Fon and other fungal contamination. To estimate in planta fungal biomass, three-week-old plants were cultivated in spore suspensions of different Fon strains with shaking (85 rpm). Samples were collected at different time points post-inoculation and qRT-PCR was conducted to determine the levels of FonOpm12 and watermelon ClRps10 (used as an internal control). Relative fungal biomass was expressed as the FonOpm12/ClRps10 ratio (Gao et al., 2022).
2.4 RNA extraction and reverse transcriptase (RT)-qPCRTotal RNA was extracted from mycelia cultured in PDB for 2 d using RNA Isolater reagent (Vazyme Biotech). First-strand cDNA was synthesized using HiScript II qRT SuperMix kit (Vazyme Biotech) following the manufacturer’s recommendations. Reactions for qPCR were prepared with 2× AceQ qPCR SYBR Green Master Mix (Vazyme Biotech) and run on a LightCycler 96 instrument (Roche). FonActin served as an internal control to normalize the qPCR data and relative expression levels of genes were calculated using the 2–△△CT method. The primers used are listed in Supplementary Table S1.
2.5 Microscopic examinationsTo observe subcellular localization, fresh mycelia and macroconidia of WT::FonKin4-GFP strain were harvested from two-day-old culture grown on potato dextrose broth (PDB) at 26°C and then examined under a Zeiss LSM780 confocal microscope using the appropriate conditions for capturing the GFP signal.
2.6 Yeast two-hybrid (Y2H) assaysY2H assays were conducted following the manufacturer’s instructions for the Matchmaker Gold Y2H System (Clontech, Mountain View, CA, United States) with the yeast strain Y2H Gold. The coding sequences of full-length FonKin4 and the N-terminal region of FonPARP1 were amplified with gene-specific primers (Supplementary Table S1) and cloned into double-cleaved pGBKT7 and pGADT7 using the principle of homologous recombination, respectively. Paired vectors were co-transformed through LiAc/SS carrier DNA/PEG method (Gietz and Schiestl, 2007). After incubation on a basic medium (SD) lacking Leu and Trp at 30°C for 5 d, the transformants were screened on SD/−Leu/−Trp/-His/−Ade medium supplemented with 40 μg/mL X-α-gal (Clontech, Mountain View, CA, United States).
2.7 Co-immunoprecipitation (Co-IP) assaysThe coding sequences of the targeted genes were incorporated into pYF11 vector, featuring a C-terminal GFP tag, and/or pHZ126 vector, harboring a 3× FLAG tag (Yin et al., 2020). Subsequently, these constructs were transformed into WT protoplasts, and the transformants were grown on PDA containing hygromycin B and/or neomycin, followed by confirmation using PCR amplification and Western blotting. For Co-IP assays, mycelia cultured in PDB for 2 d were harvested. Total proteins were extracted using a lysis buffer (1 M Tris–HCl, pH7.4, 0.5 M EDTA, 1 M NaCl, 0.1% Triton X-100, 1 mM DTT, and 1× protease inhibitor cocktail) and precipitated with GFP-Trap beads (ChromoTek, Planegg-Martinsried, Germany) following the manufacturer’s instructions. The eluted proteins were subsequently detected by immunoblotting via using either anti-GFP antibody or anti-FLAG antibody (Cat. # A8592, Sigma-Aldrich), respectively.
2.8 Purification of recombinant proteinsThe coding sequences of FonPARP1, FonKin4, and FonKin4-ST were cloned into the pGEX4T-3 vector, featuring a GST tag, or pET32a vector, possessing an HIS tag. Prokaryotic expression and purification of recombinant proteins were performed as previously described (Liu et al., 2019). Briefly, E. coli Rosetta cells carrying the recombinant vectors were induced by 1 mM Isopropyl ß-D-1-thiogalactopyranoside (IPTG; Sigma-Aldrich) at 18°C for 20 h. Subsequently, the recombinant proteins were purified using a GST fusion protein purification kit (Genscript, Piscataway, NJ, United States) and Profinity IMAC Ni-charged resin (Bio-Rad, Hercules, CA, United States), respectively.
2.9 In vitro pull-down assaysGST-tagged recombinant proteins were immobilized on glutathione-Sepharose beads (Yeasen, Shanghai, China) and then incubated with HIS-tagged recombinant proteins in GST buffer at 4°C for 3 h. The eluted proteins were then separated on a 12.5% SDS-PAGE and immunoblotted with anti-GST antibody (Cat. #A00865, GenScript) or anti-HIS antibody (Cat. #A00186, GenScript), respectively.
2.10 Western blotting assaysWestern blotting assay was carried out as previously described (Gao et al., 2022). Samples were separated on SDS-PAGE gels of varying percentages (8%, 12.5% or 15%) and subsequently transferred onto an Immobilon-P transfer membrane (Millipore, Billerica, MA, United States). The membranes were then immunoblotted with appropriate antibodies. Blot detection was accomplished using the ECL chromogenic reagent (Thermo Fisher Scientific, Waltham, MA, United States), and the bands were scanned or imaged using the Tanon automatic gel imaging system (Tianneng Corporation, Shanghai, China).
2.11 Immunoprecipitation-liquid chromatography–tandem mass spectrometry (IP-LC–MS/MS)The FonPARP1-GFP strain was cultured in PDB for 2 d, and total proteins were extracted from fresh mycelial samples using above-mentioned lysis buffer. Subsequently, proteins were precipitated with GFP-Trap beads following the manufacturer’s instructions, eluted using the elution buffer (100 mM Tris–HCl, pH7.6, 4% SDS, and 1 mM DTT), and subjected to LC–MS/MS analysis. MS/MS spectra was searched using MASCOT engine (Matrix Science, London, United Kingdom; version 2.2) against Fusarium oxysporum f. sp. lycopersici 4,287 protein sequences database (NCBI). For protein identification, the following options were used. The peptide mass tolerance was set to 20 ppm, and the MS/MS fragment tolerance was set to 0.1 Da. The enzyme was trypsin. The missed cleavage was set to 2. The ion score of peptides was set to ≥20.
2.12 In vitro phosphorylation assaysIn vitro phosphorylation assays were conducted as previously reported (Xia et al., 2021). Briefly, 2 μg HIS-FonKin4 with or without 4 μg HIS-FonPARP1 were incubated in a 50 μL kinase reaction buffer (20 mM HEPES-KOH, 10 mM MgCl2, 40 mM ATP, 25× protein inhibitor cocktail, pH7.5) at 30°C for 1 h. After incubation, the reaction was halted by adding 4× SDS loading buffer. Subsequently, the proteins were separated on 8% SDS-PAGE and detected using anti-phosphor Ser/Thr antibody (Cat. #PP2551, ECM Biosciences, Aurora, CO, United States) or anti-HIS antibody (Cat. #A00186, GenScript).
2.13 In vitro PARylation assaysIn vitro PARylation assays were performed as described previously (Kong et al., 2021; Yao et al., 2021). Briefly, 500 ng HIS-FonPARP1 were incubated in a 50 μL PARylation buffer (50 mM Tris–HCl, 50 mM NaCl, 10 mM MgCl2, pH8.0) with 0.2 mM NAD+, 1× activated DNA (BPS Bioscience, San Diego, CA, USA), and 3 μg GST-FonKin4 at 26°C for 3 h. Subsequently, the samples were separated on 8% and 12.5% SDS-PAGE. PARylated proteins were detected via immunoblotting with anti-poly-ADPR antibody (Cat. #MABE1031, Sigma-Aldrich), anti-GST antibody (Cat. #A00865, GenScript) or anti-HIS antibody (Cat. #A00186, GenScript).
For in vitro phosphorylation-mediated self-PARylation assays, 4 μg HIS-FonPARP1 underwent prior in vitro phosphorylation by HIS-FonKin4-ST, as described above. Then, 50 μL PARylation buffer was then added, followed by incubation at 26°C for 30 min. The reaction was stopped by adding 4× SDS loading buffer, and the proteins were then separated on 8% and 12.5% SDS-PAGE. Phosphorylated proteins were detected by anti-phosphor Ser/Thr antibody. PARylated proteins detected through immunoblotting with streptavidin-HRP (Cat. #21126, Thermo Fisher Scientific, Waltham, MA, USA) for Biotin NAD+ or anti-HIS antibody (Cat. #A00186, GenScript).
2.14 Phylogenetic analysisProtein sequences were searched from National Center for Biotechnology Information (NCBI) using HMMER (Hidden Markov Model search, Hmmsearch) web server as a query. Sequences were initially aligned using CLUSTALX. Subsequently, phylogenetic analysis was conducted with MEGA7 software using the neighbor-joining method based on boostrap = 1,000. SMART protein database (http://smart.embl-heidelberg.de/) was utilized to analyze protein conserved domains.
2.15 Statistical analysisAll experiments were conducted independently three times. The data were subjected to statistical analysis using either the Student’s t-test or one-way analysis of variance (ANOVA). Significant differences were defined by probability values of p < 0.05 or p < 0.01.
3 Results 3.1 Characterization of FonPARP1 and FonPARG1 in FonTo investigate the biological functions of PARylation in Fon, we initially searched for genes encoding PARylation-related PARP and PARG enzymes. HMMER was utilized to query the F. oxysporum f. sp. lycoperici reference genome database using Arabidopsis AtPARP and AtPARG protein sequences as references (Prakash et al., 2017). This analysis led to the characterization of two genes (FOXG_07574 and FOXG_05947) that potentially encode PARP and PARG enzymes in F. oxysporum (Supplementary Figure S1A), respectively. We confirmed the sequences for the predicted open reading frames (ORF) of these two genes through cloning and sequencing from Fon, subsequently naming them FonPARP1 and FonPARG1. FonPARP1 exhibited close relatedness to its orthologues in F. oxysporum f. sp. melonis, F. verticillioides, and F. graminearum but was distantly related to those in Magnaporthe oryzae, Aspergillus nidulans, and Neurospora crassa (Supplementary Figure S1B). FonPARP1, comprising 748 amino acids, featured BRCT and WGR domains at its N-terminus as well as PARP regulatory and PARP catalytic domains at its C-terminus (Supplementary Figure S1C). This structural arrangement exhibited high conservation across various fungi (Supplementary Figure S2). FonPARG1 clustered with FomPARG1 from F. oxysporum f. sp. melonis (Supplementary Figure S1D) and encompassed 476 amino acids, characterized by a single conserved PARG catalytic domain (Supplementary Figures S1A,E, S3). Overall, the conservation of FonPARP1 and FonPARG1 across multiple fungi, along with the identification of their critical structural domains, suggests their likely conservation of function.
3.2 Generation of targeted deletion mutants and complementation strains for FonPARP1 and FonPARG1To elucidate the biological functions of FonPARP1 and FonPARG1 in Fon, we generated targeted deletion mutants ΔFonPARP1 and ΔFonPARG1 employing the homologous recombination strategy (Supplementary Figures S4A,B). These deletion mutants were verified by Southern blotting using an HPH probe, confirming the presence of a single insertional HPH fragment in the mutants but not in the WT strain (Supplementary Figures S4C,D). RT-qPCR results revealed that the transcripts of FonPARP1 and FonPARG1 were barely detectable in the respective deletion mutants (Supplementary Figures S4E,F). To construct a complementation strain ΔFonPARP1-C, we expressed a native promoter-driven FonPARP1-GFP cascade in ΔFonPARP1 background (Supplementary Figure S4G). Considering the importance of glutamic acid (E) at 988 position for the enzymatic activity of human PARP1 (Altmeyer et al., 2009; Chen et al., 2019), we created a mutated variant FonPARP1E729K, in which conserved E residue was replaced with lysine (K). This variant was fused with a GFP tag and introduced into ΔFonPARP1 to generate a mutated complementation strain ΔFonPARP1-CE729K (Supplementary Figure S4G). Since various lines of the mutants and complementation strains exhibited comparable phenotypes, we selected one representative line of ΔFonPARP1, ΔFonPARG1, ΔFonPARP1-C, and ΔFonPARP1-CE729K for further investigations.
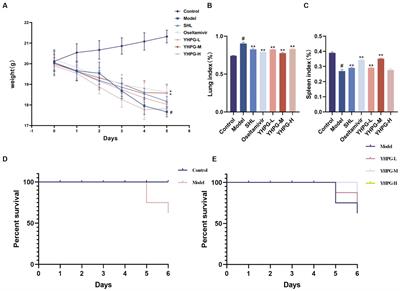
3.3 FonPARP1 is required for Fon pathogenicityWe then investigated the involvement of FonPARP1 and FonPARG1 in Fon pathogenicity by assessing the disease-causing ability of the deletion mutants ΔFonPARP1, ΔFonPARG1 as well as their complementation strains. In repeated experiments, ΔFonPARP1-inoculated plants exhibited less severe disease symptoms, with 55% displaying cotyledon yellowing and slight wilting (Figures 1A,B). The disease ratings of these plants decreased by 41% compared to WT-inoculated plants (Figures 1A,B). Conversely, ΔFonPARG1-inoculated plants showed no significant difference in disease development compared to WT-inoculated plants (Supplementary Figures S5A,B). Notably, ΔFonPARP1-CE729K-inoculated plants presented similar disease symptoms to the ΔFonPARP1-inoculated plants, and the disease index was reduced by 39% compared to WT-inoculated plants (Figures 1A,B). These data suggest that FonPARP1 is essential for Fon pathogenicity, whereby FonPARG1 is not required, and emphasize the critical role of conserved E729 amino acid in the function of FonPARP1 in Fon pathogenicity.
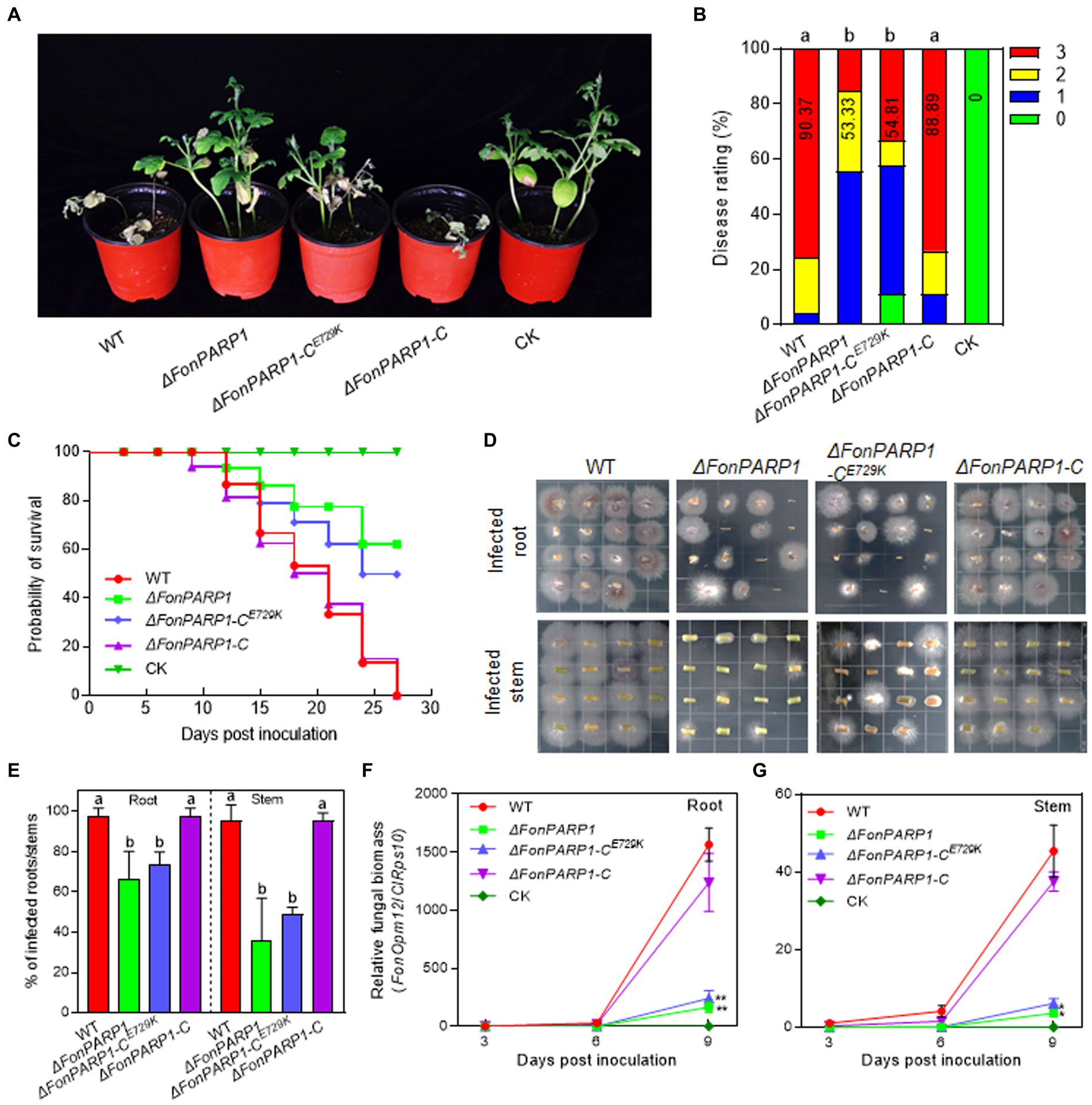
Figure 1. FonPARP1 is required for Fon pathogenicity on watermelon. (A,B) Disease phenotype (A) and disease ratings (B) of watermelon plants inoculated with WT, ΔFonPARP1, ΔFonPARP1-CE729K or ΔFonPARP1-C strains at 21 dpi. (C) Survival curves of watermelon plants inoculated with WT, ΔFonPARP1, ΔFonPARP1-CE729K or ΔFonPARP1-C strains during a 27-d experimental period. (D,E) Tissue examination (D) and percentages (E) of fungal colonies grown from roots and stems of WT-, ΔFonPARP1-, ΔFonPARP1-CE729K- or ΔFonPARP1-C-inoculated watermelon plants at 15 dpi. (F,G) Relative in planta fungal biomass of WT, ΔFonPARP1, ΔFonPARP1-CE729K, and ΔFonPARP1-C in roots and stems of the inoculated plants. Relative fungal biomass was quantified by qRT-PCR and is presented as the ratio of FonOpm12/ClRps10. Experiments were independently performed three times with similar results. Data presented in (B–G) represent the means ± SD from three independent experiments. Different letters in (B,E) or asterisks in (F,G) indicate significant differences at p < 0.05 level by one-way ANOVA or Student’s t-test, respectively.
To further confirm the function of FonPARP1 and FonPARG1 in Fon pathogenicity, we monitored disease progression in watermelon plants following inoculation with the deletion mutants and their corresponding complementation strains. We observed a delay of ~3 d in the onset of disease-caused death in plants inoculated with ΔFonPARP1 and ΔFonPARP1-CE729K compared with those inoculated with ΔFonPARP1-C, which began to die at 9 d post-inoculation (dpi) (Figure 1C). During a 27-d experiment period, the death rates progressed at a slower pace in plants inoculated with ΔFonPARP1 and ΔFonPARP1-CE729K than those inoculated with WT and ΔFonPARP1-C (Figure 1C). At 27 dpi, all WT- and ΔFonPARP1-C-inoculated plants had died, while only 38 and 50% of the ΔFonPARP1- and ΔFonPARP1-CE729K-inoculated plants succumbed to the disease, respectively (Figure 1C). In contrast, the disease progression and death rate of the ΔFonPARG1-inoculated plants were similar to those of the WT-inoculated plants (Supplementary Figure S5C), confirming that FonPARG1 is not required for Fon pathogenicity.
We conducted further examinations to evaluate the in planta fungal biomass of ΔFonPARP1 and its complementation strains, ΔFonPARP1-C and ΔFonPARP1-CE729K, in inoculated plants. In tissue examination assays, root and stem segments from ΔFonPARP1 and ΔFonPARP1-CE729K-infected plants supported fewer Fon colonies compared to segments from WT- and ΔFonPARP1-C-infected plants (Figure 1D), showing reductions of 25 ~ 31% in roots and 47 ~ 60% in stems (Figure 1E). Further, the relative in planta fungal biomass of ΔFonPARP1 and ΔFonPARP1-CE729K was obviously lower than that of WT and ΔFonPARP1-C in both roots and stems of the infected plants, displaying reductions of 85 ~ 90% in roots and 87 ~ 91% in stems compared to the WT strain at 9 dpi (Figures 1F,G). However, it is worth noting that ΔFonPARP1 still exhibited the ability to penetrate the cellophane membrane, similar to WT and ΔFonPARP1-C (Supplementary Figure S6). Taken together, these results indicate that FonPARP1 plays a vital role in Fon pathogenicity, likely by affecting the invasive growth within watermelon plants rather than influencing the penetration ability.
3.4 FonKin4 interacts with FonPARP1To explore the biochemical mechanism of FonPARP1 in regulating Fon pathogenicity, we sought to identify putative FonPARP1-interacting factors by performing LC–MS/MS characterization of FonPARP1-GFP-immunoprecipitated proteins. In this analysis, we detected 48 peptides corresponding to 53 genes that may encode putative FonPARP1-interacting factors (Supplementary Table S2). Among these, we shortlisted FOXG_01025, encoding a putative protein kinase consisting of 1,111 amino acids (Supplementary Figure S7A). Given that FOXG_01025 is very similar to yeast Kin4 (D’Aquino et al., 2005), we named it FonKin4. FonKin4 was phylogenetically clustered with homologs from other plant pathogenic filamentous fungi (Supplementary Figure S7B) and possesses a typical S_TKc domain with a conserved active threonine residue (Supplementary Figures S7C,D). To validate the direct interaction between FonPARP1 and FonKin4, we conducted Y2H and pull-down assays. Since the expression of whole PARP1 interfered with cell growth in yeast (La Ferla et al., 2015; Yao et al., 2021), we thus used the N-terminal region of FonPARP1 (FonPARP1-N) for the Y2H assays. FonKin4 exhibited interaction with the N-terminal region of FonPARP1 (Figure 2A). In pull-down assays, GST-FonKin4, but not bare GST, successfully pulled-down HIS-FonPARP1 (Figure 2B). Additionally, Co-IP assays involving strains co-expressing PARP1-GFP and FonKin4-FLAG confirmed the in vivo association between FonKin4 and FonPARP1, as FonKin4-FLAG was co-immunoprecipitated with FonPARP1-GFP (Figure 2C). Furthermore, subcellular localization assays showed that FonKin4-GFP was predominantly localized around septa in both mycelia and macroconidia of Fon (Figure 2D).
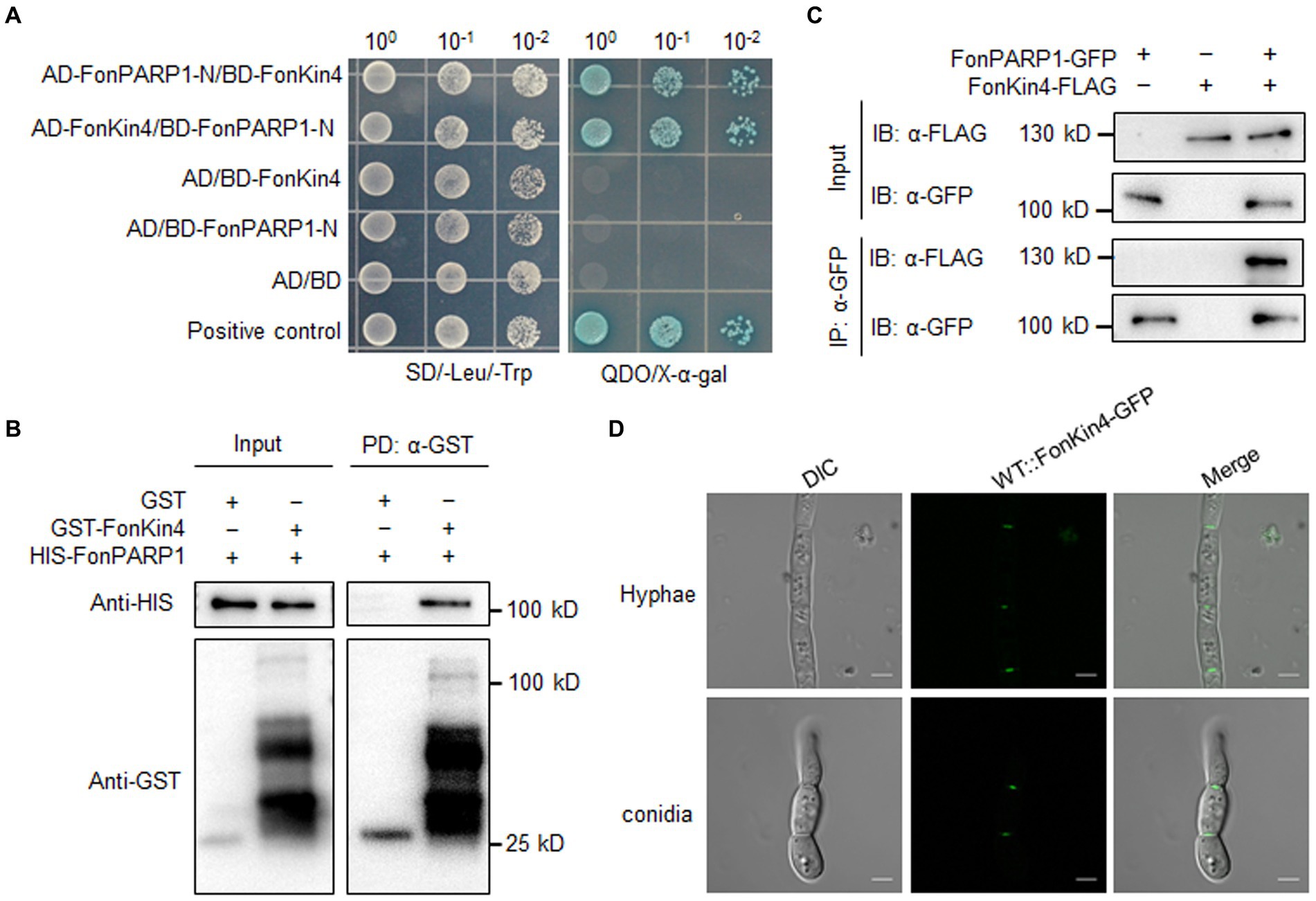
Figure 2. FonKin4 interacts with FonPARP1. (A) Interaction between FonKin4 and FonPARP1 in Y2H assays. Equal amounts of yeast cells expressing different combinations were grown on SD/−Leu/−Trp and QDO/X-α-gal plates. (B) Interaction between FonKin4 and FonPARP1 in pull-down assays. HIS-tagged FonPARP1 was incubated with immobilized GST-tagged FonKin4. The samples were detected by anti-HIS or anti-GST antibody. (C) Interaction between FonKin4 and FonPARP1 in Co-IP assays. FonPARP1-GFP and FonKin4-FLAG were co-expressed in WT strain. Total proteins were extracted, immunoprecipitated with GFP-Trap beads, and detected by anti-GFP or anti-FLAG antibody. (D) Subcellular localization of FonKin4-GFP in mycelia and macroconidia of Fon. Scale bar = 5 μm.
3.5 FonKin4 is required for Fon pathogenicityTo explore whether FonKin4 plays a role in mediating Fon pathogenicity, we created a targeted deletion mutant ΔFonKin4 (Supplementary Figure S8A). The obtained deletion mutant was validated through Southern blotting, detecting a single insertional HPH fragment, which was absent in WT (Supplementary Figure S8B). Additionally, RT-qPCR analysis revealed a significant reduction in FonKin4 transcript level in the mutant (Supplementary Figure S8C). Concurrently, we generated a complementation strain ΔFonKin4-C by expressing a native promoter-driven FonKin4-GFP fusion in ΔFonKin4 (Supplementary Figure S8D). Notable, the threonine (T) residue at 209 position in the activation T-loop is crucial for the enzymatic activity of ScKin4 (D’Aquino et al., 2005; Caydasi et al., 2014). We thus created a kinase-dead variant FonKin4T462A, in which T462, corresponding to T209 in ScKin4, was substituted with alanine (A), and introduced it into ΔFonKin4, resulting in a complementation strain ΔFonKin4-CT462A, harboring a FonKinT462A-GFP fusion (Supplementary Figure S8D).
To explore the involvement of FonKin4 in Fon pathogenicity, we evaluated the disease-causing ability of the deletion mutant ΔFonKin4 and its complementation strain. In repeated experiments, ΔFonKin4-inoculated plants displayed milder disease symptoms, with 11% remaining healthy and 56% exhibiting yellow cotyledons at 21 dpi (Figures 3A,B). Compared to the disease severity observed in WT-inoculated plants, the disease rating of ΔFonKin4-inoculated plants decreased by 55% (Figures 3A,B), while ΔFonKin4-C-inoculated plants exhibited comparable disease levels to WT-inoculated plants (Figures 3A,B). Notably, ΔFonKin4-CT462A-inoculated plants exhibited similar disease levels to those of ΔFonKin4-inoculated plants, leading to a 51% reduction in the disease index compared to WT-inoculated plants (Figures 3A,B). In disease progress monitoring experiments, we observed a delay of ~6 d in the onset of disease-induced mortality in ΔFonKin4-inoculated plants compared to WT- and ΔFonKin4-C-inoculated plants (Figure 3C). The death rates progressed more slowly in ΔFonKin4- and ΔFonKin4-CT462A-inoculated plants compared to WT- and ΔFonKin4-C-inoculated plants (Figure 3C). At 27 dpi, all WT- and ΔFonKin4-C-inoculated plants had succumbed to the disease, while ~72% and ~ 61% of ΔFonKin4- and ΔFonKin4-CT462A-inoculated plants remained alive, respectively (Figure 3C).
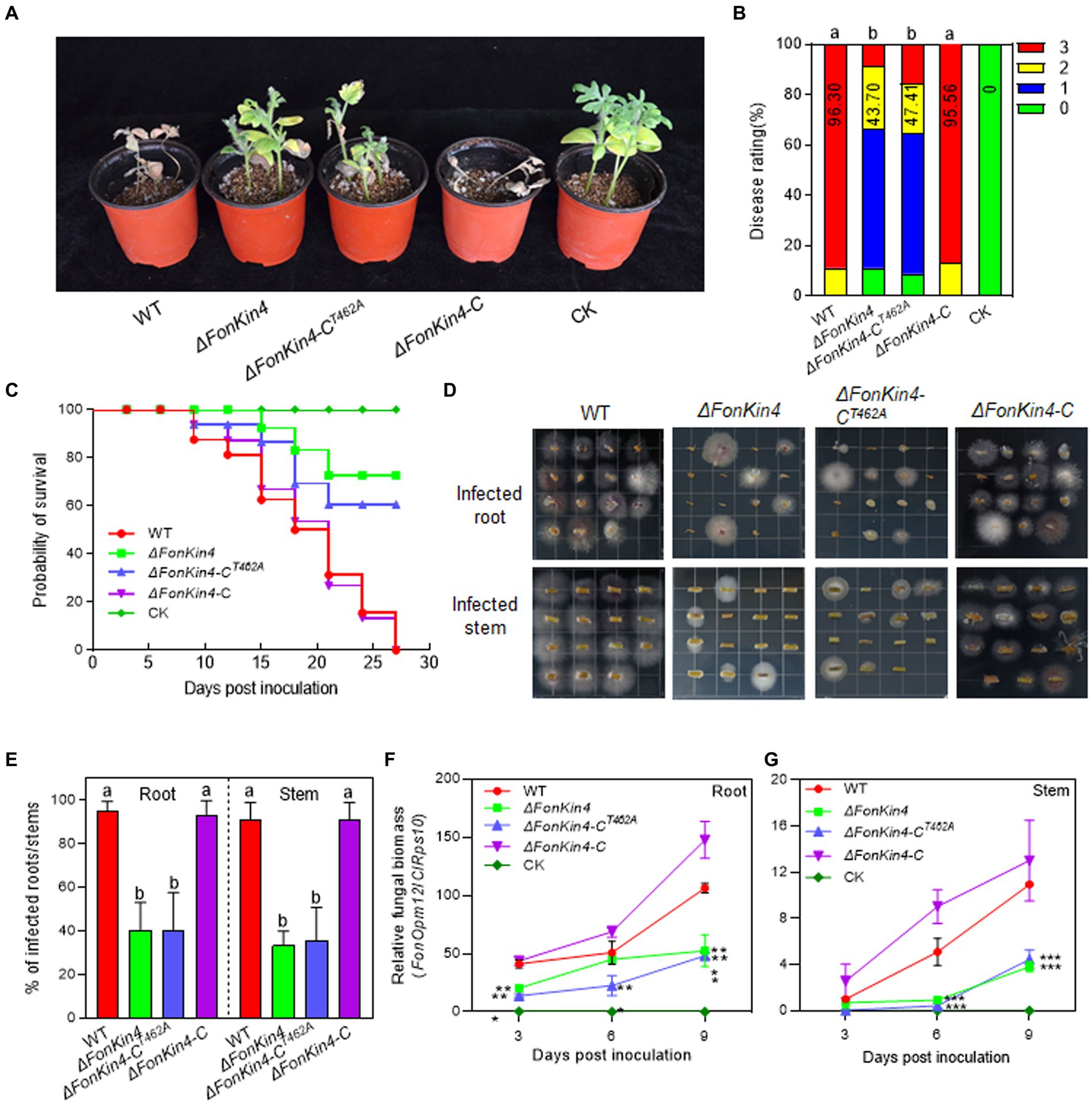
Figure 3. FonKin4 is required for Fon pathogenicity on watermelon. (A,B) Disease phenotype (A) and disease ratings (B) of watermelon plants inoculated with WT, ΔFonKin4, ΔFonKin4-CT462A or ΔFonKin4-C strains at 21 dpi. (C) Survival curves of watermelon plants inoculated with WT, ΔFonKin4, ΔFonKin4-CT462A or ΔFonKin4-C strains during a 27-d experimental period. (D,E) Tissue examination (D) and percentages (E) of fungal colonies grown from roots and stems of WT, ΔFonKin4, ΔFonKin4-CT462A or ΔFonKin4-C-inoculated watermelon plants at 15 dpi. (F,G) Relative in planta fungal biomass of WT, ΔFonKin4, ΔFonKin4-CT462A or ΔFonKin4-C in roots and stems of the inoculated plants. Relative fungal biomass was quantified by qPCR and is presented as the ratio of FonOpm12/ClRps10. Experiments were independently performed three times with similar results. Data presented in (B–G) represent the means ± SD from three independent experiments. Different letters in (B,E) or asterisks in (F,G) indicate significant differences at p < 0.05 level by one-way ANOVA or Student’s t-test, respectively.
In tissue examination assays, root and stem segments from the ΔFonKin4- and ΔFonKin4-CT462A-infected plants displayed reduced Fon colonies compared to those from the WT- and ΔFonKin4-C-infected plants (Figure 3D), showing reductions of 54 ~ 55% in roots and 55 ~ 58% in stems (Figure 3E). Moreover, the relative in planta fungal biomass of ΔFonKin4 and ΔFonKin4-CT462 was significantly lower compared to the WT and ΔFonKin4-C in both roots and stems of the infected plants. Specifically, the relative in planta fungal biomass decreased by 51% ~ 55% in roots and 59 ~ 65% in stems of the ΔFonKin4- and ΔFonKin4-CT462-inoculated plants compared to the WT-inoculated ones at 9 dpi (Figures 3F,G). Interestingly, the penetration ability of ΔFonKin4 through the cellophane membrane was similar to that of the WT and ΔFonKin4-C (Supplementary Figure S6). Collectively, these results indicate that, similar to its interacting partner FonPARP1, FonKin4 plays a functional role in Fon pathogenicity by affecting the invasive growth within watermelon plants rather than influencing the penetration ability, and the conserved T462 residue is critical for FonKin4 function in Fon pathogenicity.
3.6 FonKin4 and FonPARP1 are involved in the basic biological processes of FonWe also investigated whether FonPARP1, FonPARG1, and FonKin4 are involved in the basic biological processes of Fon. In repeated experiments, ΔFonPARG1 showed similar phenotypes to WT regarding mycelial growth, conidiation, spore germination, and macroconidia morphology (Figures 4A–F), indicating that FonPARG1 is not involved in these basic biological processes of Fon. Moreover, ΔFonPARP1 displayed slower growth on MM than WT and ΔFonPARP1-C (Figures 4A,B), while remained no changes in growth on PDA or other basic biological processes of Fon (Figures 4A–F). However, ΔFonKin4 and ΔFonKin4-CT462A exhibited slower growth on both PDA or MM (Figures 4A,B) and produced fewer macroconidia, with a reduction of ~40% compared to WT and ΔFonKin4-C (Figure 4C). The macroconidia produced by ΔFonKin4 and ΔFonKin4-CT462A germinated normally (Figure 4C) but displayed abnormal morphology with fewer septa and shorter lengths than those of WT and ΔFonKin4-C (Figures 4D–F). Particularly, ~80% of the ΔFonKin4- and ΔFonKin4-CT462A-produced macroconidia were shorter than 20 μm and had at most 1 septum, whereas >85% of macroconidia produced by WT and ΔFonKin4-C were longer than 20 μm and had more than 2 septa (Figures 4D–F). These results suggest that FonKin4 and its intact kinase activity play key roles in regulating vegetative growth, conidiation, and conidial morphology of Fon, while FonPARP1 affects mycelial growth under nutrient-scarce conditions.
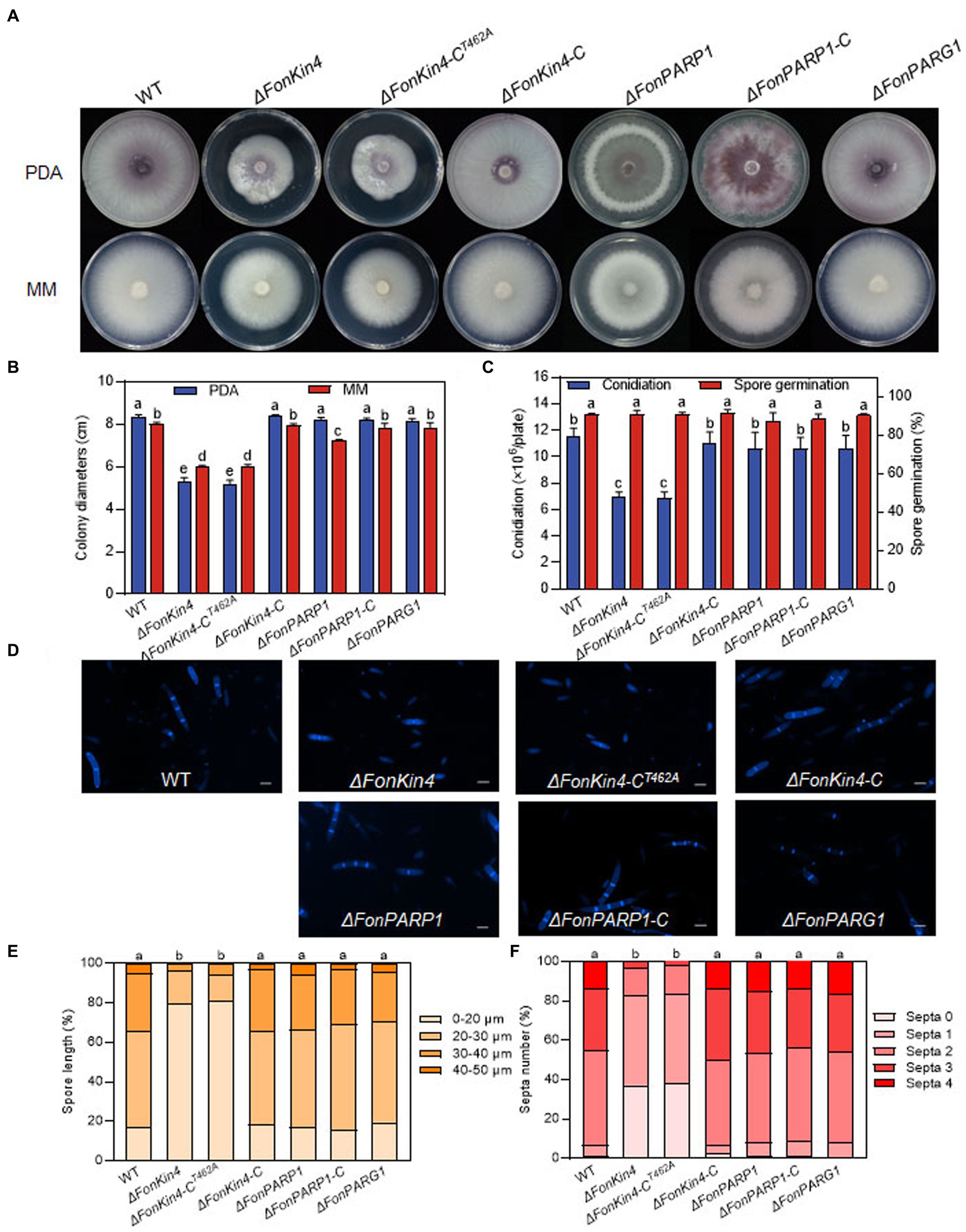
Figure 4. FonKin4 and FonPARP1 are involved in regulating the basic biological processes of Fon. (A,B) Mycelial growth (A) and colony diameters (B) of different strains grown on PDA and MM plates at 7 d. (C) Conidiation and spore germination of different strains. (D–F) Morphology (D), length (E), and septum numbers (F) of macroconidia produced by different strains. Scale bar = 5 μm. Experiments were independently performed three times with similar results. Data presented in (B,C,E,F), represent the means ± SD from three independent experiments, and different letters indicate significant differences (p < 0.05, one-way ANOVA).
3.7 FonPARP1, FonPARG1, and FonKin4 are involved in the abiotic stress responses of FonPrevious studies have revealed that PARP is engaged in responses to various abiotic stresses in mice and Arabidopsis (Shieh et al., 1998; De Block et al., 2005; Vanderauwera et al., 2007; Luo and Kraus, 2012). Therefore, we examined the involvement of FonPARP1, FonPARG1, and FonKin4 in the stress responses of Fon by comparing the mycelial growth of the deletion mutants, complementation strains, and WT on PDA supplemented with various stress-inducing agents. Overall, ΔFonPARP1, ΔFonPARP1-CE729K, ΔFonPARG1, ΔFonKin4, and ΔFonKin4-CT462A exhibit differential responses to tested stress-inducing agents compared to WT, ΔFonPARP1-C, and ΔFonKin4-C (Figures 5A,B). In response to cell wall perturbing reagents, ΔFonPARP1 and ΔFonPARP1-CE729K showed increased sensitivity to Congo red (CR) and sodium dodecyl sulfate (SDS) but enhanced tolerance to Calcofluor white (CFW), while ΔFonPARG1 displayed higher sensitivity to SDS but increased resistance to CR and CFW compared to WT (Figures 5A,B). Concerning oxidative and osmotic stresses, ΔFonPARP1 and ΔFonPARP1-CE729K exhibited higher tolerance to H2O2, paraquat, and NaCl but became more vulnerable to CaCl2 compared to WT (Figures 5A,B). ΔFonPARG1 was more tolerant to H2O2 and NaCl but more sensitive to CaCl2 (Figures 5A,B). Interestingly, ΔFonKin4 and ΔFonKin4-CT462A exhibited enhanced tolerance to all tested stress-inducing agents (Figures 5A,B). Collectively, these data indicate that FonPARP1, FonPARG1, and FonKin4 have similar functions in oxidative stress responses while playing distinct roles in responses to osmotic and cell wall perturbing stresses.
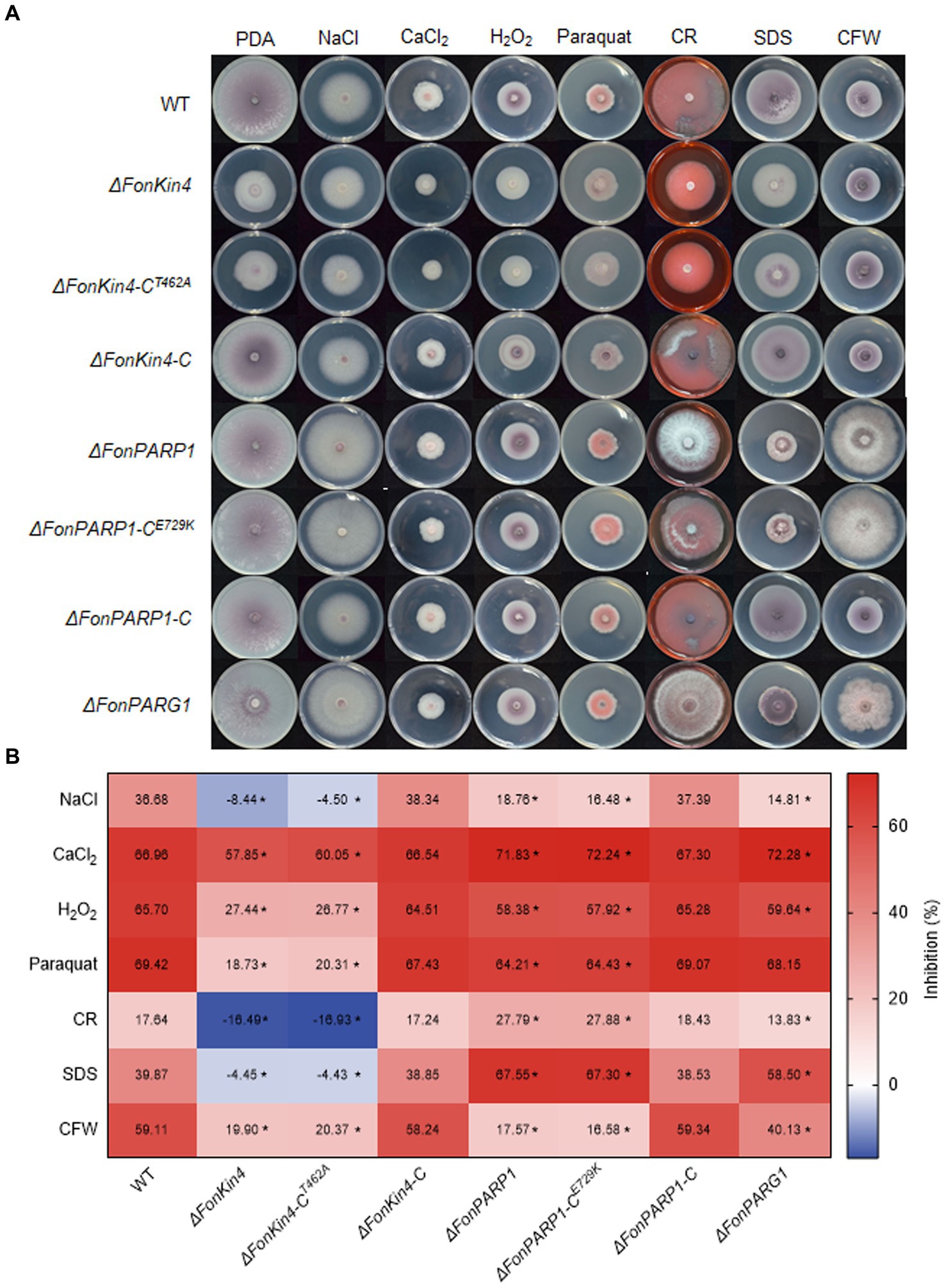
Figure 5. FonPARP1, FonPARG1, and FonKin4 are involved in the abiotic stress responses in Fon. (A) Mycelial growth and (B) inhibition rates of WT, ΔFonKin4, ΔFonKin4-CT462A, ΔFonKin4-C, ΔFonPARP1, ΔFonPARP1-CE729K, ΔFonPARP1-C, and ΔFonPARG1 on PDA supplemented with different agents at 7 d. Experiments were independently performed three times with similar results. Data presented in (B) represent the means ± SD from three independent experiments, and asterisks indicate significant differences (p < 0.05, Student’s t-test).
3.8 FonKin4 is a Ser/Thr protein kinase whose enzymatic activity is sufficient for its pathogenicity functionIt was previously shown that ScKin4 possesses protein kinase activity (Pereira and Schiebel, 2005). As FonKin4 contains a typical S_TKc domain (Figure 6A) and shares 53% identity with ScKin4 (Supplementary Figure S7B), we assessed its kinase activity using an in vitro phosphorylation assay. The recombinant GST-FonKin4 exhibited self-phosphorylation at Ser/Thr site(s), detected using an anti-phospho Ser/Thr antibody (Figure 6B), confirming FonKin4 as a protein kinase. To explore the importance of the S_TKc domain in FonKin4, we generated a truncated variant FonKin4-ST, containing the S_TKc domain (Figure 6A). We purified the recombinant HIS-FonKin4-ST protein, which also exhibited self-phosphorylation activity in the in vitro phosphorylation assay (Figure 6C). To investigate the significance of the specific residue T462 within the S_TKc domain, we mutated the conserved T residue to A in both GST-FonKin4 and HIS-FonKin4-ST. Surprisingly, neither GST-FonKin4T462A nor HIS-FonKin4-STT462A exhibited self-phosphorylation activity in the in vitro phosphorylation assay (Figures 6B,C). These results suggest that FonKin4 is an active Ser/Thr protein kinase, and the S_TKc domain, along with the conserved T462, is essential for FonKin4 kinase activity.

Figure 6. FonKin4 is a Ser/Thr protein kinase and its S_TKc domain is sufficient for its pathogenicity function in Fon. (A) Schematic representation of the structure of FonKin4 protein. (B,C) Intact FonKin4 (B) and truncated S_TKc domain-containing fragment FonKin4-ST (C) show Ser/Thr protein kinase activity detected by anti-phosphor Ser/Thr antibody. (D,E) Disease phenotype (D) and disease ratings (E) of watermelon plants inoculated with WT, ΔFonKin4 or ΔFonKin4-CST strains at 21 dpi. Experiments were independently performed three times with similar results. Results from one representative experiment are shown in (D). Data presented in (E) represent the means ± SD from three independent experiments, and different letters indicate significant differences from WT (p < 0.05, one-way ANOVA).
To further assess the role of the FonKin4-ST in the pathogenicity function of FonKin4, we generated a complementation strain ΔFonKin4-CST by introducing a native promoter-driven FonKin4-ST construct into ΔFonKin4. Pathogenicity tests revealed that the disease symptoms and rating of the ΔFonKin4-CST-inoculated plants were comparable to those of WT-inoculated plants (Figures 6D,E). At 21 dpi, 78% of the plants in both the WT- and ΔFonKin4-CST-inoculated groups had succumbed to the disease. In contrast, only 24% of the plants in the ΔFonKin4-inoculated group were affected (Figure 6E). These results provide evidence that FonKin4-ST restores the pathogenicity defect in ΔFonKin4, highlighting the essential role of enzymatic activity in the pathogenicity function of FonKin4 in Fon.
3.9 FonKin4 Phosphorylates FonPARP1 to enhance its activityThe interaction between FonPARP1 and FonKin4 led us to speculate whether FonPARP1 PARylates FonKin4 or FonKin4 phosphorylates FonPARP1. To investigate this, we first examined whether FonPARP1 PARylates FonKin4 in vitro. Since GST-FonKin4 is about 130 kD in size, which is close to HIS-FonPARP1, in vitro PARylation assays with short and long exposures were conducted to distinguish possible PARylated GST-FonKin4 band from PARylated HIS-FonPARP1 band. No self-PARylated HIS-FonPARP1 band was observed in the absence of NAD+ (Figure 7A, lane 6). However, when NAD+ was added, it resulted in a clear self-PARylated HIS-FonPARP1 band, especially a smear with longer size of the PARylated product in long exposure, indicating that HIS-FonPARP1 was enzymatically active (Figure 7A, lane 7). In contrast, no self-PARylated HIS-FonPARP1E729K band was seen in the presence of NAD+ (Figure 7A, lanes 4 and 5), indicating that mutation of E729 completely abolished the enzymatic activity of FonPARP1. On the other hand, we did not observe any PARylated GST-FonKin4 band, even in long exposure, when GST-FonKin4, HIS-FonPARP1, and NAD+ were added to the reaction (Figure 7A, lane 8), revealing that FonPARP1 does not PARylate FonKin4 in vitro. Next, we explored the possibility of FonKin4-mediated FonPARP1 phosphorylation through in vitro phosphorylation assays. In the presence of HIS-FonKin4-ST, we observed a phosphorylated HIS-FonPARP1 band using an anti-phosphor Ser/Thr antibody (Figure 7B, lane 4). However, no visible band was detected in the reaction with HIS-FonKin4-STT462A (Figure 7B, lane 5), indicating the function of FonKin4 in phosphorylating FonPARP1 at Ser/Thr site(s).
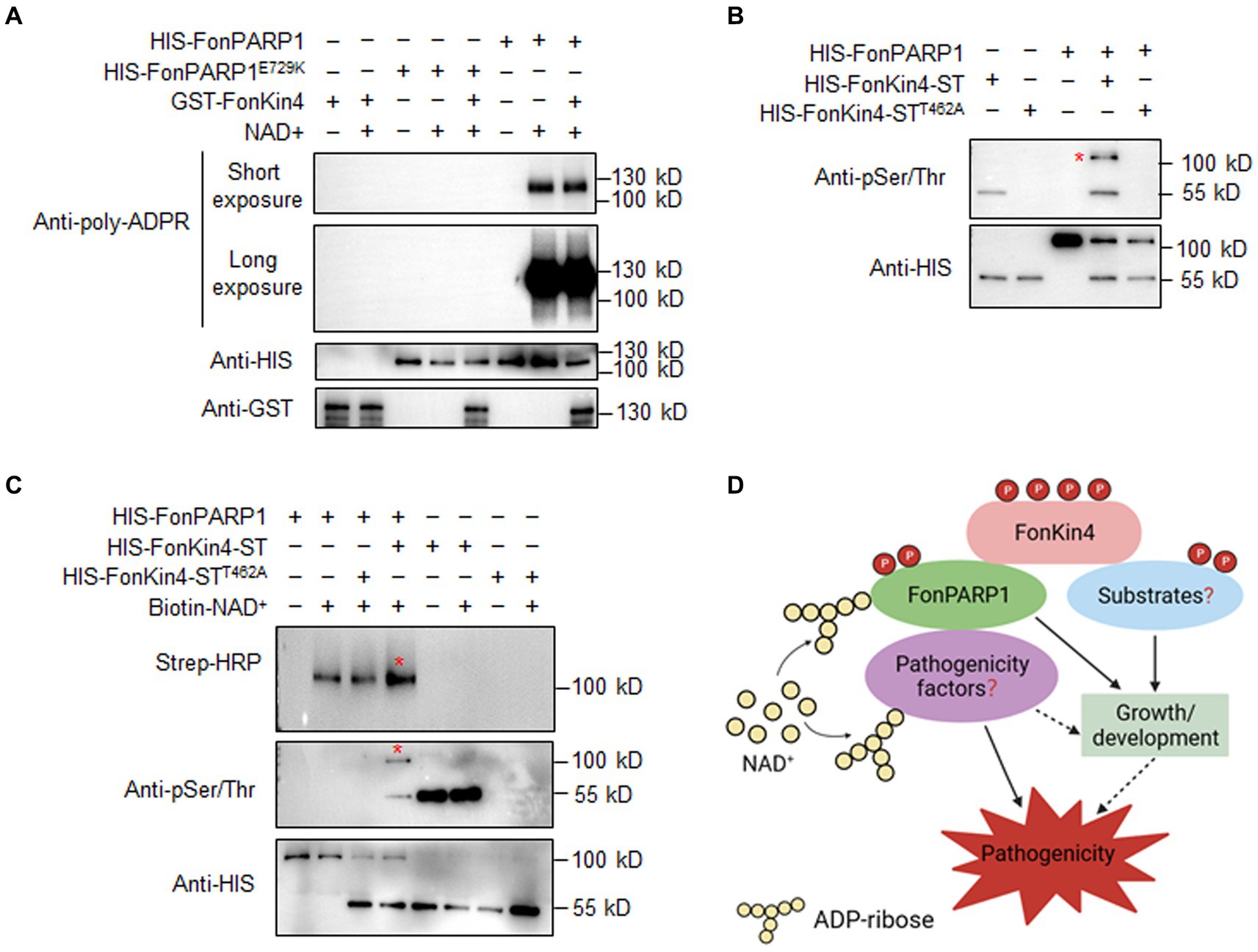
Figure 7. FonKin4 phosphorylates FonPARP1 to enhance its enzymatic activity and a proposed model illustrating the functions of the FonKin4-FonPARP1 cascade in Fon pathogenicity. (A) FonPARP1 possesses self-PARylation activity but does not PARylate FonKin4 in vitro. Short exposure, 1 min; long exposure, 5 min. (B) FonKin4 phosphorylates FonPARP1 in vitro. (C) FonKin4-mediated phosphorylation enhances the self-PARylation activity of FonPARP1 in vitro. Experiments were independently performed three times with similar results. Red asterisks in (B) and (C) indicate the self-PARylated and phosphorylated FonPARP1, respectively. (D) A proposed working model deciphering the functions of the FonKin4-FonPARP1 cascade in Fon pathogenicity.
To determine the functional significance of FonKin4-mediated phosphorylation of FonPARP1, we assayed the enzymatic activity of FonPARP1 by examining its self-PARylation level after being phosphorylated by FonKin4. In the presence of biotin-NAD+, the self-PARylation of HIS-FonPARP1 was clearly detected by streptavidin-HRP (Figure 7C, lane 2 in upper panel), which was also distinguishable in the presence of HIS-FonKin4-STT462A (Figure 7C, lanes 3 in upper panel). However, when HIS-FonKin4-ST was added to the reaction, we observed a phosphorylated band of HIS-FonPARP1 (Figure 7C, lane 4 in middle panel) and enhanced self-PARylation of HIS-FonPARP1 (Figure 7C, lane 4 in upper panel) compared to the reactions without HIS-FonKin4-ST or with HIS-FonKin4-STT462A (Figure 7C, lanes 2 and 3 in upper panel). These data indicate that FonKin4-mediated phosphorylation facilitates the activity of FonPARP1.
4 DiscussionProtein PARylation, catalyzed by PARPs and mainly degraded by PARGs, has been implicated in various biological processes in mammals and plants (Gibson and Kraus, 2012; Feng et al., 2
留言 (0)