
記住我








The global emergence of multidrug-resistant Gram-negative bacteria is the most challenging clinical and public health problem (Hernando-Amado et al., 2019; Antimicrobial Resistance Collaborators, 2022). Despite significant progress in biomedical research, many untreatable infectious diseases are considered the leading causes of human death worldwide. Nosocomial and community-acquired infections caused by opportunistic Gram-negative pathogens are becoming increasingly difficult to treat as both intrinsic and acquired antibiotic resistance has increased significantly in recent years (Theuretzbacher et al., 2020).
Among Gram-negative opportunistic pathogens, Stenotrophomonas maltophilia has been the subject of an increased interest and extensive research over the last two decades. The number of reported S. maltophilia infections has considerably risen and the bacterium has been classified as the most common Gram-negative carbapenem-resistant pathogen in patients with bacteremia in some US hospitals (Cai et al., 2020).
Stenotrophomonas maltophilia is a globally dispersed, non-fermenting Gram-negative bacillus frequently isolated in the environment, particularly from water sources, soil, sediment, plants, and animal specimens (Aznar et al., 1992; Nakatsu et al., 1995; Jakobi et al., 1996; Berg et al., 1999; Johnson et al., 2003; Ivanov et al., 2005; Romanenko et al., 2008; Berg, 2009). According to the List of Prokaryotic Names with Standing in Nomenclature, the genus Stenotrophomonas comprises at least 25 validated species exhibiting great genetic diversity and metabolic heterogeneity both within the Stenotrophomonas genus and within a single species (Ryan et al., 2009; Turrientes et al., 2010; Pompilio et al., 2016). Comprehensive taxonomic and phylogenomic studies have shown that S. maltophilia includes multiple cryptic species, forming the Stenotrophomonas maltophilia complex (Smc) and a distinction that defies conventional classification approaches (Ochoa-Sánchez and Vinuesa, 2017; Kumar et al., 2020; Singh et al., 2023). Identification of phylogenetic relationships among Stenotrophomonas spp. based on analysis of core protein sequences revealed 24 species-level clades of Smc (Gröschel et al., 2020; Li et al., 2024).
The bacterium demonstrates high adaptability to various environments, including nutrient-limited and hostile conditions. S. maltophilia is capable of utilizing a wide range of carbon sources such as trichloroethylene, toluene, chloroform, glucose, and benzene (Lee et al., 2002; Pompilio et al., 2011; Mukherjee and Roy, 2013).
Stenotrophomonas maltophilia is an opportunistic pathogen intrinsically resistant to multiple and broad-spectrum antibiotics. The bacterium is associated with a number of serious diseases and contributes significantly to the pathogenesis of multibacterial infections. S. maltophilia causes infections in various human organs, including the respiratory, gastrointestinal, and urinary tracts. It can cause severe pneumonia, catheter-associated bacteremia/septicemia, osteochondritis, mastoiditis, meningitis, and endocarditis (Denton and Kerr, 1998; Brooke, 2012; Chang et al., 2015). The bacterium is frequently recovered in the lungs of cystic fibrosis (CF) patients; according to various studies, the frequency ranges from 10% to 30% (Waters et al., 2011; Cuthbertson et al., 2020). During the global COVID-19 pandemic, S. maltophilia has been recognized as one of the most common causative agents of respiratory co-infections and bacteremia in critically ill COVID-19 patients. Furthermore, S. maltophilia isolates detected in sputum samples obtained from these patients had the highest rates of multidrug resistance among other bacteria infecting COVID-19 patients (Yang S. et al., 2021; Ishikawa et al., 2022; Langford et al., 2023).
Stenotrophomonas maltophilia is also of interest as an active member of polymicrobial bacterial communities: it can influence the metabolism of neighboring microorganisms, either through antagonistic suppression of other species or by symbiotic coexistence. A vivid example of such inter-species communication can be observed in CF patients, where S. maltophilia colonizes the host along with other major pathogens, such as Pseudomonas aeruginosa, Staphylococcus aureus, nontuberculous mycobacteria, or Burkholderia cenocepacia (Goss et al., 2004; Coutinho et al., 2008).
The pathogenesis of infections caused by S. maltophilia is determined by numerous virulence factors (VFs), molecules that facilitate bacterial colonization of the host at the cellular level, thereby initiating the infectious process. S. maltophilia possesses a considerable spectrum of VFs or putative factors associated with virulence. These factors include surface cell-associated structures (lipopolysaccharides, type IV pili, flagella, fimbriae, and nonpilus adhesins); the production of a wide spectrum of extracellular enzymes (e.g., proteases, esterases, lipases), hemolysin, siderophores, and cytotoxins; the ability to form biofilms on abiotic surfaces and host tissues; SmeYZ, SmeDEF, SbiAB, and MacABCsm efflux pumps; and the secretion of small molecules in the environment via Quorum Sensing (QS) intercellular communication system [the diffusible signal factor (DSF) and outer membrane vesicles (OMV); Looney, 2005; Brooke, 2012; Ferrer-Navarro et al., 2013; Abbott and Peleg, 2015; Trifonova and Strateva, 2019; Wu C.-J. et al., 2022].
In this review, we briefly summarize the current knowledge on S. maltophilia virulence and provide an overview of the literature introducing the virulence determinants and their regulation in S. maltophilia. We have limited the scope of the review to virulence, partially touching upon inter-species bacterial interactions and iron uptake systems in the context of virulence, and have not referred to antibiotic resistance.
Adhesins as virulence factorsAdherence of the bacterium to host tissues is a crucial step in the host-pathogen interaction. At this stage, the pathogen attached to the host cell initiates its own biochemical processes aimed at its proliferation, invasion of host cells, secretion of toxic molecules, and activation of host cell signaling cascades.
Bacterial adherence factors, also known as adhesins, are polypeptides or polysaccharides. Protein adhesins are cell-surface components or appendages that can be divided into two groups: fimbrial and afimbrial. Polysaccharide adhesins are generally associated with the bacterial cell wall, outer membrane, or capsule. It should be noted that adhesion functions in pathogenesis are not limited to the initial host-pathogen interaction; adhesins also play a significant role in subsequent stages of infection (see below). To provide a holistic view of the role of various VFs in the development of S. maltophilia infection, cell-associated and extracellular VFs are summarized and illustrated in Figure 1.
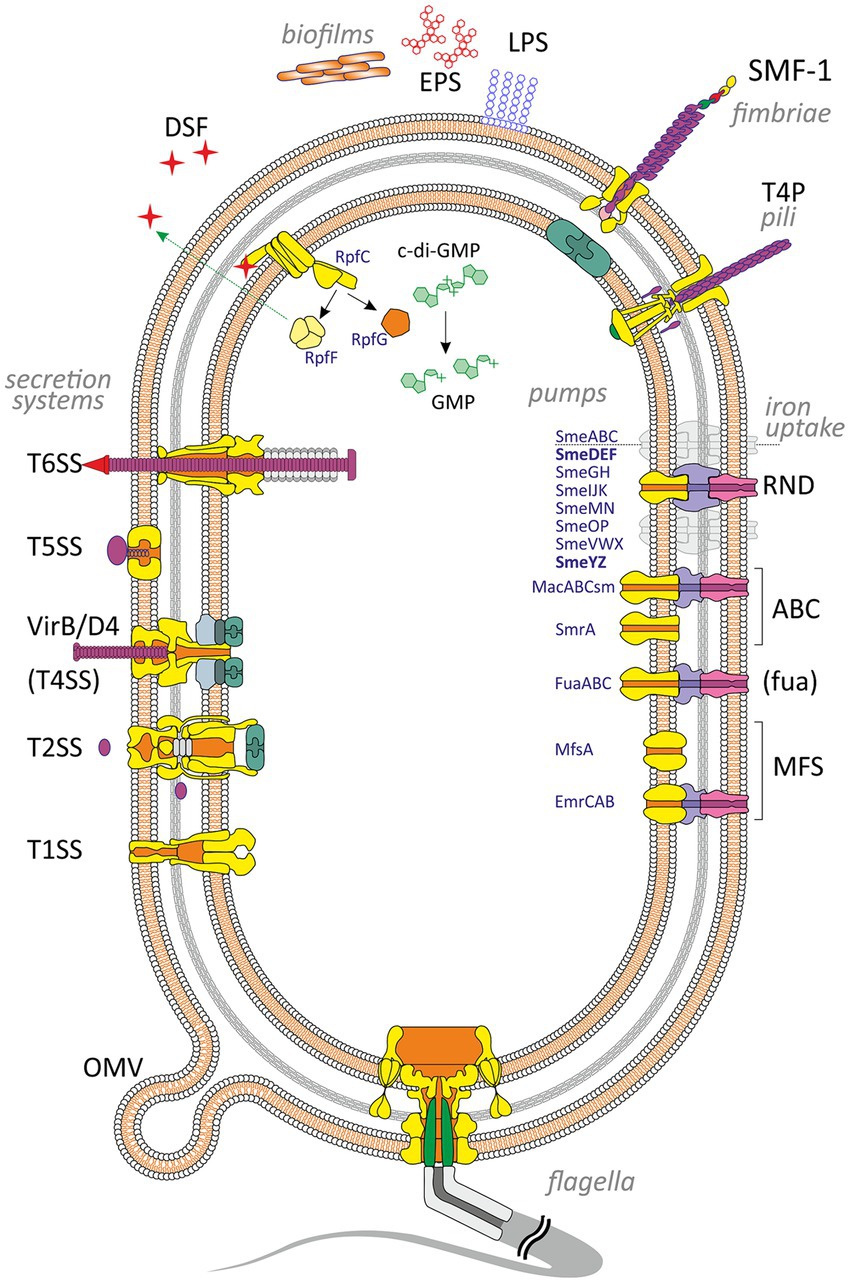
Figure 1. Virulence and putative virulence factors in Stenotrophomonas maltophilia. Surface cell-associated structures include lipopolysaccharides (LPS), type IV pili (T4P), flagella, fimbriae (SMF-1), and non-pilus adhesins (not shown). Extracellular enzymes are secreted through type I, II, IV, V, and VI secretion systems. Small molecules efflux to the environment via the diffusible signal factor (DSF) and outer membrane vesicles (OMV). S. maltophilia produces extracellular polymeric substances and forms a self-secreted polymeric matrix, biofilms, consisting of exopolysaccharides (EPS), DNA, and proteins. The intracellular c-di-GMP level contributes to numerous virulence factors (see the text for details). Different types of efflux pumps revealed in S. maltophilia are shown on the right. The pumps involved in virulence are marked in bold.
Cell-surface polysaccharidesStenotrophomonas maltophilia has lipopolysaccharides (LPS) comprising lipid A, core oligosaccharide, and O-antigen (Neal and Wilkinson, 1982; Jucker et al., 1996; Zhang and Kong, 2002; McKay et al., 2003). LPS is a robust inducer of TNF-α production by macrophages due to its lipid A moiety, that has been elegantly demonstrated by Waters et al. (2007) in a mouse model. Despite the relatively weak invasiveness of S. maltophilia, the level of TNF-α production after stimulating cells of macrophage cell line RAW for 4 h with purified lipid A isolated from S. maltophilia was significantly higher than the corresponding level obtained after stimulation with lipid A from the laboratory P. aeruginosa PAO1 control strain (Waters et al., 2007).
Core oligosaccharides play an essential role in LPS formation and therefore in virulence. Defects in core oligosaccharides of certain bacteria (e.g., P. aeruginosa, Bordetella bronchiseptica) are associated with decreased virulence or the emergence of avirulent strains (Goldberg et al., 1995; West et al., 2000).
The contribution of O-antigens to virulence has also been reported to be significant for many bacteria. Bacteria lacking O-antigens or possessing defective antigen molecules due to disruptions in biosynthesis may exhibit decreased virulence properties. This phenomenon has been demonstrated, in particular, for the species Burkholderia pseudomallei, P. aeruginosa, and B. abortus (Goldberg et al., 1995; DeShazer et al., 1998; Ugalde et al., 2000). In S. maltophilia, defective LPS lacking the O-antigen can affect biofilm production, twitching motility (the ability to move on surfaces using type IV pili), and swimming motility (Huang et al., 2006; Brooke et al., 2008). Stenotrophomonas maltophilia lipopolysaccharides show structural diversity between strains within the genus, with at least 31 different O-antigens (Winn and Wilkinson, 2001; Waters et al., 2007).
The metabolic process of carbohydrates and their incorporation into LPS in S. maltophilia is regulated by several genes. The spgM gene involved in this process encodes a bifunctional enzyme with both phosphoglucomutase and phosphomannomutase activity. This gene is homologous to algC which controls alginate and LPS biosynthesis in P. aeruginosa (McKay et al., 2003; Flores-Treviño et al., 2019). McKay et al. (2003) have shown that spgM mutants with shorter O-polysaccharide chains lost virulence in the rat lung model and exhibited increased susceptibility to complement-mediated killing.
Two operons, rmlBACD and xanAB, are involved in the synthesis of polysaccharides in S. maltophilia. Huang et al. (2006) have conducted an SDS-PAGE analysis of purified LPS from S. maltophilia rmlA, rmlC, and xanB mutants and found these genes necessary for LPS O-antigen biosynthesis. In addition, xanB is required for the production of the LPS core region. The authors also suggested that rml and xanB are involved in the biosynthesis of exopolysaccharides (EPS): the rmlA and rmlC mutants exhibited decreased biofilm production on polystyrene and increased biofilm density on glass. Meanwhile, the xanB mutant displayed lower biofilm production only on polystyrene. Besides contributing to biofilm formation, alterations in LPS caused by the rmlAC and xanB mutations, may lead to changes in outer membrane appendages, such as flagella and type IV pili, thereby interfering with motility and attachment (Huang et al., 2006).
FlagellaStenotrophomonas maltophilia possesses one or multiple polar flagella that confer swimming motility, swarming and chemotaxis. Flagella facilitate primary adherence to biotic and abiotic surfaces, contribute to colonization and invasion in the early stages of infection, and trigger specific immune responses by host cells (de Oliveira-Garcia et al., 2002; Huang et al., 2006; Pompilio et al., 2010, 2018; Zgair and Chhibber, 2011).
Wu et al. have identified three flagellar genes, fliC1, fliC2, and fliC3 which form an operon in the sequenced S. maltophilia genome. The authors generated single, double, and triple mutants corresponding to these genes and revealed that each gene contributed to swimming, adhesion, and biofilm formation. The ability to attach, swim, and form biofilms decreased proportionally to the number of deactivated genes, with the triple mutant losing its swimming ability and significantly compromising adhesion and biofilm formation. Thus, flagella in motile pathogens can be considered important VFs (Moens and Vanderleyden, 1996; Duan et al., 2013; Wu C.-J. et al., 2022).
Pompilio et al. (2010) have constructed two S. maltophilia mutants in which the fliI gene was inactivated. The highly conserved fliI gene encodes a substrate-specific ATPase (FliI) that provides energy for the active translocation of flagellar structural components in a wide range of bacterial species (Yonekura et al., 2002; Di Bonaventura et al., 2007). These two flagellum-deficient S. maltophilia fliI mutants exhibited decreased adherence to CF-derived bronchial epithelial IB3-1 cells and compromised swimming motility (Pompilio et al., 2010).
Inbred BALB/c mice (commonly used as animal models for drug and vaccine testing) pretreated intranasally with purified S. maltophilia flagellin and instilled with S. maltophilia 4 h later exhibited significantly increased levels of pro-inflammatory cytokines IL-1β and TNF-α, myeloperoxidase activity, caspase-1 activity, and nitric oxide compared to control groups (Zgair and Chhibber, 2012). The pretreated mice also demonstrated elevated levels of neutrophils, lymphocytes, and monocytes in their bronchoalveolar lavage fluid providing nonspecific protection for the animals against S. maltophilia as well as S. aureus infections (Zgair and Chhibber, 2012). In another study, Pompilio et al. (2018) compared the severity of disease caused by aerosol challenge of mice of the DBA/2 N strain with the clinical isolate S. maltophilia SM111 and its aflagellate isogenic mutant (ΔfliI). Pompilio et al. (2018) did not observe a significant trend in body weight changes, pulmonary persistence, lung damage, or mortality in mice infected with wild-type and fliI− strains. The authors suggested that flagella and motility might not represent S. maltophilia virulence traits involved in the pathogenesis of lung infection. Meanwhile, the expression of TNF-α in murine lungs infected with the aflagellate mutant was significantly reduced (Pompilio et al., 2018).
Hypothetically, in the context of a chronic infection, it is admissible that a bacterium lacking flagellin as a significant immunogenic factor may gain a survival advantage diminishing the host’s immune response. This hypothesis could be corroborated by observations in patients with CF: a substantial portion of P. aeruginosa isolates (39%) from chronically colonized patients were nonmotile and resistant to phagocytosis by macrophages (Mahenthiralingam et al., 1994). Nevertheless, the majority of studies have reported a positive correlation between motility and primary adhesion (e.g., de Oliveira-Garcia et al., 2002; Waters et al., 2007; Pompilio et al., 2010; Zgair and Chhibber, 2011; Madi et al., 2016). Perhaps, the aforementioned speculation that flagella do not contribute to S. maltophilia virulence may be relevant only to the later (chronic) stage of the disease when the initial adhesion step preceding the infection has already occurred.
The motility of S. maltophilia and the level of flagellin gene expression depend on environmental conditions and are tightly controlled by comprehensive genetic systems that have not been fully investigated. In particular, cyclic diguanosine monophosphate (c-di-GMP), the ubiquitous second messenger has been recognized as an important intracellular signaling molecule involved in the regulation of flagellin gene expression in many bacteria. c-di-GMP is also implicated in controlling various bacterial physiological processes, including the cell cycle, adherence, motility, the production of VFs, and biofilm formation (Ross et al., 1987; Hengge, 2009).
The intracellular concentration of c-di-GMP is modulated by two groups of enzymes with opposing activities, diguanylate cyclases (DGCs) and phosphodiesterases (PDEs). DGCs contain a conserved GGDEF domain and synthesize c-di-GMP by condensation of two GTP molecules. PDEs, with either EAL or HD-GYP domains, degrade c-di-GMP into linear guanosine dinucleotide (pGpG) or guanosine monophosphate (GMP) (Chan et al., 2004; Christen et al., 2005, 2006; Caly et al., 2014). An increased level of c-di-GMP, resulting from the action of DGCs, is associated with a sessile lifestyle and biofilm formation which is relevant to chronic bacterial infections. Conversely, lower concentrations of c-di-GMP due to phosphodiesterase activity, are found in motile bacteria during acute infection processes (Hengge, 2009; Moscoso et al., 2011; Cheng et al., 2019). In S. maltophilia, the mechanisms by which c-di-GMP controls flagellar synthesis and flagella numbers are still poorly understood, and there is a paucity of studies focused on the molecular basis of its functioning.
In some bacteria, the expression of flagellar genes is activated and controlled by specific genetic determinants known as master regulators and their homologs, e.g., FlrA (FleQ) in P. aeruginosa and Vibrio cholerae, flaK and flaM in Vibrio parahaemolyticus (Ritchings et al., 1995; Stewart and McCarter, 1996; Arora et al., 1997; Klose and Mekalanos, 1998; Hickman and Harwood, 2008).
Yang et al. (2014) reported that flagellar gene expression in S. maltophilia is controlled by FleQ (Smlt2295). This transcriptional regulator, an enhancer-binding protein, is homologous to the similar master regulator in P. aeruginosa and it is the major target for the c-di-GMP produced by the wrinkly spreader phenotype (Wsp) chemosensory system pathway (Hickman et al., 2005; Yang et al., 2014). FleQ works together with a putative ATPase, FleN, and is inhibited by binding c-di-GMP. Inhibition of this complex and, therefore, Wsp pathway activation results in elevated expression of biofilm-associated pel, psl, and cdr operons and a reduction of flagellar gene expression (Hickman et al., 2005; Hickman and Harwood, 2008; Yang et al., 2014). The situation is reversed if the concentration of c-di-GMP is low: FleQ remains unbounded, and it leads to increased expression of flagellar genes, and, therefore, the ability of bacteria to be sessile in biofilms decreases.
Liu et al. (2017) have reported a correlation between increased expression of bsmR which encodes an eponymous regulatory protein, an EAL domain-containing PDE, and increased bacterial swimming and decreased cell aggregation in S. maltophilia CGMCC 1.1788. Therefore, bsmR is suggested to be a negative regulator of biofilm formation and a positive regulator of swimming motility. The bsmR operon controls the expression of at least 349 genes, of which 34 are involved in flagellar synthesis and are under positive regulation of the FsnR transcription factor (Liu et al., 2017). BsmR degrades c-di-GMP to activate the expression of FsnR. This flagellar-assembly-related transcription factor binds directly to the promoter regions of two operons, Smlt2303 and Smlt2318, initiating the transcription of flagella-associated genes (Kang et al., 2015; Zheng et al., 2016).
Zhang et al. (2022) analyzed genes potentially affecting the c-di-GMP level in S. maltophilia, specifically those encoding proteins containing GGDEF, EAL, and HD-GYP domains. The authors identified 33 putative c-di-GMP turnover enzymes in the genome of S. maltophilia using the Simple Modular Architecture Research Tool (SMART) (Letunic and Bork, 2018) and constructed mutants of all 33 genes via insertional inactivation. Among the mutants analyzed, 12 bacterial strains exhibited a deficiency in swimming motility while one showed promotion, that suggests the 13 corresponding genes may contribute to the regulation of bacterial swimming motility. The authors also made an important observation that the mutation-induced degeneration or inactivation of DGCs or PDEs do not necessarily alter the cellular c-di-GMP level and bacterial swimming motility. Therefore, further investigations are needed to assess the contribution of each gene to swimming motility.
Among all the enzymes analyzed, the authors also identified and characterized a novel Fe2+-dependent phosphodiesterase named SisP (S. maltophilia iron-sensing PDE). SisP increased its activity and facilitated bacterial swimming upon stimulation with ferrous iron in a dose-dependent manner, and the degradation of c-di-GMP led to FsnR-dependent transcription of flagellar genes (Zhang et al., 2022).
Fimbriae and piliType 1 fimbriae (SMF-1) are an important VF that confers to S. maltophilia the ability to adhere to various specific host epithelia (de Oliveira-Garcia et al., 2002; Zgair and Chhibber, 2011; Giltner et al., 2012). In particular, it has been reported that adherence to biotic (epithelial cells) and abiotic surfaces (such as medical implants and catheters) was inhibited by anti-SMF-1 antibodies (de Oliveira-Garcia et al., 2003). Fimbriae are also involved in early stages of biofilm formation (de Oliveira-Garcia et al., 2003) and can agglutinate red blood cells (Crossman et al., 2008). Antibodies against SMF-1 fimbriae, but not preimmune serum, inhibited hemagglutination in a dose-dependent manner (de Oliveira-Garcia et al., 2003).
It is worth noting that SMF-1 fimbriae were revealed in all clinical isolates (n = 46) obtained from patients (de Oliveira-Garcia et al., 2003), whereas S. maltophilia strains isolated from environmental sources did not possess them (Nicoletti et al., 2011). Therefore, fimbriae are thought to be the significant structures involved in the adhesion and colonization of the lung epithelium.
Fimbrial protein production is controlled by the Smf-1 fimbrial operon, which includes Smlt0706-Smlt0709 (Crossman et al., 2008). This type of fimbriae is assembled by the bacterial chaperone-usher pathway (Thanassi et al., 1998). Despite S. maltophilia fimbrin is closely related to fimbrial adhesins of most members of the Enterobacteriaceae and the CupA fimbriae of P. aeruginosa, the N-terminal amino acid sequences of S. maltophilia Smf-1 significantly differ from those belonging to other families of fimbriae (ranging from 50% to 61%). This suggests that this family of fimbriae may extend to other genetically distant non-enteric bacterial genera (Vallet et al., 2001; de Oliveira-Garcia et al., 2003).
Type IV pili (T4P) are considered a significant VF associated with twitching motility, adhesion to biotic and abiotic surfaces, colonization, and biofilm formation in various bacterial pathogens (Doig et al., 1988; Saiman et al., 1990; Giltner et al., 2012). T4P have also been reported to mediate P. aeruginosa virulence through interdependent action with the type III secretion system (T3SS), thereby promoting its effector injection into the host cell (Heiniger et al., 2010; Shikata et al., 2016).
To date, the role of type IV pili in S. maltophilia virulence has not been sufficiently studied. Kalidasan and Neela (2020) have proposed a theoretical model for S. maltophilia type IV pilus based on Rapid Annotations using Subsystem Technology (RAST) analysis that provided high quality genome annotations for bacterial genomes across the whole phylogenetic tree, and previous reports on P. aeruginosa.
Extensive sequence variation in the type IV pilin adhesion precursor gene has been revealed by Fluit et al. (2022). Meanwhile, no significant correlations have been reported between virulence and the presence of the pil gene family which is involved in pilus formation. An analysis of clinical and environmental S. maltophilia strains performed by Cruz-Córdova et al. (2020) showed that the pilU gene frequencies were high enough but comparable in both groups analyzed, regardless of origin. An increase in biofilm biomass formed by CF isolates with elevated swimming and twitching motility has been reported by Pompilio et al. (2011) and, notably, the phenomenon was observed only in CF isolates. Taken together, there appears to be no direct evidence for T4P as a significant VF in S. maltophilia, and further studies are needed to clarify their contribution to its virulence.
Secretion systems and extracellular enzymesClinical S. maltophilia strains produce a variety of VFs, including proteases (StmPr1, StmPr2, StmPr3, StmPr4), lipases (lipase and phospholipase C and D), nucleases, gelatinases, elastase, esterases, hyaluronidases, fibrinolysin/streptokinase, heparinases, hemolysins, siderophores, and cytotoxins (Windhorst et al., 2002; Travassos et al., 2004; Trifonova and Strateva, 2019). These VFs contribute to bacterial colonization/persistence, induce cytotoxic and morphological effects on host cells, and play roles in various stages of the infection process (Karaba et al., 2013; DuMont et al., 2015).
Of the 11 known bacterial secretion systems (including outer membrane vesicles, OMVs), S. maltophilia possesses type I, II, IV, V, and VI secretion systems that have been identified through genome sequencing (Crossman et al., 2008; Rocco et al., 2009; Zhu et al., 2012; Adamek et al., 2014; Alavi et al., 2014). Albeit the role of these systems in virulence formation is well understood in many bacteria, only three types of S. maltophilia secretion systems (II, IV, and VI) have been described in detail.
The genome of S. maltophilia clinical strain K279a has two unlinked loci that are predicted to encode the double membrane-spanning type II secretion system, T2SS (GSP and XPS). Each locus contains 11 T2SS genes, corresponding to the core T2SS components (Karaba et al., 2013). The S. maltophilia type II secretion system mediates the secretion of at least seven protein effectors and three proteolytic activities. Proteolytic enzymes, particularly the serine proteases StmPr1, StmPr2, and StmPr3, are secreted in an XPS-dependent manner and induce structural and viability changes in lung epithelial cells, promoting the degradation of collagen, fibrinogen, fibronectin, and interleukin 8 (IL-8) (Karaba et al., 2013; DuMont et al., 2015; DuMont and Cianciotto, 2017). Another serine protease, StmPR4, has also been reported in the S. maltophilia genome (Windhorst et al., 2002; Ribitsch et al., 2012). It is thought that StmPR3, together with StmPR1 and StmPR2, contributes to the protease-mediated dysfunction of the innate immune system in cystic fibrosis (Molloy et al., 2019).
Lee et al. (2022) purified and identified a serine colistin-degrading protease (Cdp) in S. maltophilia strain Col1. Isolated from the soil, this strain exhibited high-level resistance against colistin (MIC value of 32 mg/L). Coculture and coinfection assays revealed that S. maltophilia strain Col1, bearing the cdp gene, could inactivate colistin, thereby protecting susceptible P. aeruginosa. Using colistin against P. aeruginosa infection in Drosophila melanogaster increased fly survival by 41%. In contrast, coinfection of flies with S. maltophilia strains carrying the cdp gene, did not increase the survival rate after colistin treatment. The authors noted that S. maltophilia genomes contain genes orthologous to cdp, located in a region immediately adjacent to the T2SS gene cluster (Lee et al., 2022). Thus, the colistin-degrading protease may play an important role in collective resistance to colistin in polymicrobial infections such as CF (Lee et al., 2022).
A type IV secretion system (T4SS) has been identified in the genome of both clinical and environmental S. maltophilia isolates (Nas et al., 2019). In S. maltophilia, the T4SS called the VirB/D4 system, is highly conserved within the genus and it is most similar to the T4SS of the Xanthomonas genus (Nas et al., 2019). T4SS typically comprises 12 proteins (VirB1-VirB11, and VirD4; Christie et al., 2014; Ghosal et al., 2017; Grohmann et al., 2018) and facilitates the delivery of DNA and/or protein effectors into bacterial or eukaryotic targets in a contact-dependent manner (Gonzalez-Rivera et al., 2016; Grohmann et al., 2018). The VirB10 protein, as part of the periplasm-outer membrane-spanning subcomplex, and the ATPase coupling protein VirD4 are essential for the antibacterial activity of the T4SS in S. maltophilia K279a (Bayer-Santos et al., 2019; Nas et al., 2019, 2021). The contribution of the VirB/D4 system to interspecific antagonism was elegantly demonstrated by Nas et al. (2019). They reported that S. maltophilia was capable of killing P. aeruginosa environmental strain 7700 and clinical isolates PAO1 and PAK when cocultured. Interestingly, S. maltophilia exhibited selectivity when acting on different species of heterologous bacteria, e.g., it killed Pseudomonas mendocina but not P. fluorescens, P. putida, or P. stutzeri (Nas et al., 2019). Based on studies that highlight the contribution of the type VI secretion systems in various bacteria to interbacterial killing (Basler et al., 2012; Ma et al., 2014; Ahmad et al., 2019), the authors suggested that the S. maltophilia VirB/D4 T4SS effectors are akin to those secreted by other type VI secretion systems, e.g., lipases, peptidases, nucleases, and muramidases (Ho et al., 2014).
Nas et al. (2021) identified 13 putative cognate immunity proteins in S. maltophilia that typically provide self-protection to the organism encoding the T4SS, and studied the effect of their expression in heterologous bacteria. Using these proteins, the authors revealed two potential antibacterial effectors, RS14245 and RS14255, that were required for the ability of S. maltophilia to kill heterologous bacteria, especially laboratory E. coli and clinical strains of P. aeruginosa isolated from the lungs of CF patients. The putative lipases, RS14245 and RS14255, when bound by cognate immunity proteins, did not exhibit antibacterial activity; and S. maltophilia complemented mutants lacking RS14245 and RS14255 significantly reduced their antibacterial properties (Nas et al., 2021).
The authors’ findings are intriguing from various perspectives. On the one hand, the secretion of effectors that suppress other bacterial species can be regarded as a significant VF of S. maltophilia. On the other hand, the isolation and study of such effectors hold the potential to develop novel antimicrobial drugs for the targeted therapy of infections caused by Pseudomonas and Escherichia.
Apart from providing a competitive advantage to S. maltophilia in polymicrobial communities, probably by increasing its fitness, the VirB/D4 T4SS effectors have another essential function: they inhibit apoptosis in infected lung epithelial cells but induce apoptosis in infected macrophages (Nas et al., 2019).
The Type VI Secretion System (T6SS) is a protein secretion nanomachine utilized by Gram-negative bacteria to deliver toxic effectors into target cells in a contact-dependent manner (Mougous et al., 2006; Perault et al., 2020; Crisan and Goldberg, 2022). Protein effectors exert their toxicity on the bacterial cell envelope and can degrade the peptidoglycan layer and lipid membranes, form pores and interfere with protein synthesis in the cytoplasm of competitor bacteria (Russell et al., 2011, 2013; Ahmad et al., 2019; Nolan et al., 2021). Additionally, T6SS effectors hinder host cell functions, facilitate immune evasion, thereby promoting a successful infection (Hachani et al., 2016), and participate in bacterial metal uptake by assisting low- and high-affinity transport systems in scavenging metal ions from the environment (Wang et al., 2015; Stubbendieck and Straight, 2016; Lin et al., 2017; Si et al., 2017; Han et al., 2019; Yang X. et al., 2021; Li et al., 2022).
Although T6SS genes have been identified in some S. maltophilia strains early (Alavi et al., 2014; Bayer-Santos et al., 2019), there is a paucity of experimental evidence demonstrating the function of T6SS effectors in the bacterium. Crisan et al. (2023) reported that the S. maltophilia STEN00241 clinical isolate possesses an active T6SS under standard laboratory conditions and the T6SS contributes to the elimination of some heterologous bacterial species. In particular, STEN00241 killed Burkholderia cenocepacia strain K56-2 and E. coli DH5α in a T6SS-dependent manner, but not P. aeruginosa PA14 laboratory strain, the P. aeruginosa CF isolate (PA32), and the S. aureus JE2. This selectivity in the mode of interspecific interaction within multi-species communities (elimination, competitive co-existence, or hypothetical symbiosis) suggests that the T6SS secretory function is also regulated by various environmental factors. The putative T6SS secretion triggers may be signals generated by the QS of the neighboring bacteria or their various metabolites (Lesic et al., 2009; Lin et al., 2017).
Concluding the chapter on S. maltophilia secretion systems, at least one intriguing question remains unanswered: what is the benefit to the bacterium of using both VirB/D4 T4SS and T6SS to produce functionally similar antibacterial proteins? Suggesting that it is not redundancy, the functions of these effectors and/or their trigger mechanisms are thought to be different and need to be further investigated.
To summarize, it should be noted that although five types of secretion systems (type I, II, IV, V, and VI) have been revealed in S. maltophilia genomes, further research is needed to fully comprehend their functional roles and potential interactions between the systems.
BiofilmsThe ability of S. maltophilia to form biofilms on abiotic surfaces and host tissues is an important VF that plays a crucial role in HAI and multibacterial infections and dramatically decreases the therapeutic efficacy of important antibiotics, including aminoglycosides, fluoroquinolones, and tetracycline (Di Bonaventura et al., 2004, 2007; Pompilio et al., 2010; Sun et al., 2016). Biofilms provide protection to the members of bacterial communities from exposure to antibiotics by reducing their diffusion (Tseng et al., 2013) and increasing their inactivation (Amanatidou et al., 2019). Besides, the biofilm polymer matrix gives bacteria protection from various forms of environmental stress, such as dehydration, UV exposure, salinity, and toxic metals (Hall-Stoodley et al., 2004). High cell density within biofilms and increased oxidative stress result in an elevated mutation rate and enhanced horizontal gene transfer (HGT) (Driffield et al., 2008). Compared to their planktonic counterparts, bacteria in biofilms exhibit greater resistance to nutrient starvation, pH fluctuations, and oxygen radicals (Jefferson, 2004). Biofilms may also increase the level of resistance by altering the expression of pre-existing antibiotic resistance genes (ARGs) (Høiby et al., 2010) as well as the proportion of tolerant or persister cells within the population due to a reduction in bacterial metabolic activity within the biofilm interior (Walters et al., 2003; Wood et al., 2013).
The formation of persister cells is also hypothetically possible due to a reduction in antibiotic concentration within biofilms, since it has been demonstrated that sub-MIC (minimum inhibitory concentration) levels of various antibiotics can induce persister cell formation (Dörr et al., 2009; Johnson and Levin, 2013; Kwan et al., 2013). Nutrient limitation within biofilms perhaps also affects the bacterial stringent response, where (p)ppGpp (alarmone) levels lead to slower bacterial growth and promote the formation of persister cells (McCall et al., 2019; Ro et al., 2021). In addition, biofilms protect bacteria from the host’s immune response by acting as a physical barrier, helping bacteria avoid detection and phagocytosis, and by activating response regulators, genetic switches, or suppressors that affect the activity of immune cells (Hall-Stoodley et al., 2004; González et al., 2018).
The initial stage of the biofilm formation process occurs within the first 30–60 min when planktonic cells adhere to a surface through weak and reversible interactions mediated by semiflexible fimbriae and flagella filaments. The second stage typically begins 4 h later, during which bacterial cells irreversibly attach to and colonize a surface using flagella, pili, and other surface appendages. After adhering to a surface, the cells initiate the production of extracellular polymeric substances, thereby forming a self-secreted polymer matrix of exopolysaccharides, DNA, and proteins (Flores-Treviño et al., 2019). The first microcolonies are generated by the aggregation of cells after approximately 10 h. The third stage occurs in 18–24 h when the biofilm turns into a mature phase. A mature biofilm possesses microchannels to transport water, nutrients, and debris; and bacterial cells within the biofilm express specific genes involved in QS (see below), EPS, and protein production. In mature biofilms, individual or clustered biofilm cells can detach, disperse, and colonize new niches within less than 24 h (de Kievit, 2009; Sun et al., 2016; Flores-Treviño et al., 2019).
At least several putative genes related to S. maltophilia biofilm formation have been identified. These biofilm-associated genes include: spgM (a biofunctional enzyme with phosphoglucomutase and phosphomannomutase activity); rmlA (glucose-1-phosphate thymidylyltransferase); rmlC (an epimerase RmlC, also named RfbC); xanB (a bifunctional enzyme, phosphomannose isomerase-GDP-mannose pyrophosphorylase); and rpfF (cis-11-methyl-2-dodecenoic acid, or synthase for the diffusible signal factor, DSF) (Köplin et al., 1992; Di Bonaventura et al., 2004; Huang et al., 2006; Pompilio et al., 2011; Zhuo et al., 2014). In addition to the genes listed above, numerous genes associated with the synthesis of LPS, fimbria, flagella, and pili, as well as the intracellular c-di-GMP level contribute to biofilm formation (see the corresponding chapters). For instance, almost all (30/31; 97%) S. maltophilia isolates harboring smf-1, which encodes the fimbrial protein Smf-1, were able to form biofilms (Gallo et al., 2016). The macABCsm and smeYZ genes, encoding pumps, have also been identified as essential for biofilm formation (Huang et al., 2006; Lin Y.-T. et al., 2014; Lin et al., 2015). An extended list of genes potentially associated with biofilm production can be found in the review by Flores-Treviño et al. (2019).
Recently, Strateva et al. (2023) analyzed 220 S. maltophilia strong biofilm producers and found the overall frequency of three biofilm-associated genes as follows: spgM—98.6%, rmlA—86%, and rpfF—66.5%. Meanwhile, Zhuo et al. (2014) have noted that, although the rmlA, spgM, or rpfF are closely related to biofilm formation, they do not significantly affect the average amount of biofilm.
Ramos-Hegazy et al. (2020) analyzed a transposon mutant library for mutations leading to altered biofilm formation. The authors identified the gpmA gene, which encodes a glycolytic enzyme, phosphoglycerate mutase, mediating the initial stages of S. maltophilia attachment to abiotic surfaces as well as immortalized CF-derived bronchial epithelial (CFBE) cells. The S. maltophilia isogenic mutant ΔgpmA exhibited a significant decrease in initial attachment and early biofilm formation on polystyrene plates compared to the wild type within the first 2–4 h. Interestingly, after 6 h, there was no difference in biofilm formation between the wild and mutant strains, suggesting that gpmA is involved only in the early phase of adhesion and biofilm formation (Ramos-Hegazy et al., 2020; Di Bonaventura et al., 2023).
Pompilio et al. (2020) have analyzed 85 S. maltophilia strains isolated from patients with CF and other infections and revealed that over 88% of the isolates were able to form biofilm, with non-CF strains being significantly more efficient compared to CF strains. Meanwhile, the prevalence of the multidrug-resistant phenotype was higher in CF isolates in contrast to non-CF ones (90% vs. 67%). S. maltophilia strains susceptible to piperacillin/tazobactam or meropenem produced significantly increased biofilm biomass compared to resistant strains. The authors suggested that susceptible bacteria may utilize biofilms as an alternative defense strategy to evade antibiotic action and to survive within the host (Pompilio et al., 2020).
Liu et al. (2017) have demonstrated the role of a regulatory protein mentioned above, BsmR, an EAL-domain-containing phosphodiesterase, in controlling biofilm formation and swimming motility in S. maltophilia. An increase in BsmR expression led to a significant increase in bacterial swimming motility and a decrease in cell aggregation. Thus, BsmR was identified as a negative regulator of biofilm development that degrades c-di-GMP through its EAL domain, thereby activating the expression of a transcriptional regulator, FsnR (see above), which positively controls the transcription of flagellar genes involved in swimming motility (Liu et al., 2017; Zhang et al., 2022).
An outer membrane protein, Ax21, secreted within OMVs and associated with a VF related to QS, is also implicated in biofilm formation (Ferrer-Navarro et al., 2013; An and Tang, 2018). Deletion of ax21 (Smlt0387) has been shown to reduce motility, biofilm formation, virulence to larvae of Galleria mellonella, tolerance to tobramycin, as well as alter the expression of some genes associated with virulence or antibiotic resistance (Ferrer-Navarro et al., 2013; An and Tang, 2018).
Most interestingly, the analysis of transcriptome profiles of seven clinical S. maltophilia isolates, combined with differential gene expression of biofilm vs. planktonic cells, revealed that a relatively small set of shared and commonly regulated genes is involved in the biofilm lifestyle: only about 9.5% of all genes were differentially regulated. On average, approximately 7.5% of all genes were upregulated, and about 2% of all genes were downregulated in biofilms compared to planktonic cells (Alio et al., 2020).
A comprehensive analysis of all available data on the role of various factors in the transition of bacterial cells from a planktonic to a sessile lifestyle in biofilms shows that this transformation is initiated and regulated by many mechanisms that require further study.
Efflux pumps and virulenceHistorically, efflux pumps have been considered to be among the mechanisms that provide bacteria with resistance to antimicrobials. Efflux pumps significantly contribute to the intrinsic antimicrobial resistance of S. maltophilia. However, as noted and discussed below, some types of efflux pumps possess an extended range of functions beyond the scope of “antibiotic resistance”, and these pumps are involved in the molecular mechanisms of bacterial virulence.
The genome of S. maltophilia contains a formidable arsenal of pumps belonging to various families. This includes ATP-binding cassette (ABC) pumps, MacABCsm (Lin C.-W. et al., 2014) and SmrA (Al-Hamad et al., 2009); Major Facilitator family (MFS) pumps, EmrCAB (Huang et al., 2013) and MfsA (Srijaruskul et al., 2015; Dulyayangkul et al., 2016); a fusaric acid efflux pump, FuaABC (Hu et al., 2012); and eight Resistance Nodulation Division (RND) pumps [SmeABC (Li et al., 2002), SmeDEF (Alonso and Martínez, 2000; Zhang et al., 2001; García-León et al., 2014), SmeGH (Blanco et al., 2019; Li et al., 2019), SmeIJK (Huang et al., 2014), SmeMN (Crossman et al., 2008), SmeOP (Lin C.-W. et al., 2014), SmeVWX (Chen et al., 2011; García-León et al., 2015), and SmeYZ (Lin et al., 2015)].
The efflux pumps encoded in the S. maltophilia genome are involved in the removal of a wide spectrum of toxic substances, including antibiotics. The ABC multidrug efflux pump, SmrA, contributes to the elimination of fluoroquinolones, tetracycline, and doxorubicin (Al-Hamad et al., 2009) while MacABCsm, another member of the same family, removes aminoglycosides, macrolides, and polymyxins (Lin C.-W. et al., 2014). The MFS efflux pump, EmrCABsm, facilitates the removal of nalidixic acid, erythromycin, carbonyl cyanide 3-chlorophenylhydrazone, and tetrachlorosalicylanilide (Huang et al., 2013). The fusaric acid tripartite efflux pump, FusA, is involved in the elimination of fusaric acid (Hu et al., 2012). The role of seven out of the eight RND efflux pumps (SmeABC, SmeDEF, SmeGH, SmeIJK, SmeOP, SmeVWX, and SmeYZ) in the antibiotic resistance has been also identified, except for SmeMN (Gil-Gil et al., 2020). Additionally, some RND pumps are involved in the efflux of chloramphenicol, tetracycline, macrolides, quinolones, sulfamethoxazole, trimethoprim, and trimethoprim–sulfamethoxazole (Alonso and Martínez, 2000; Sánchez and Martínez, 2018; Wu et al., 2019). The contribution of pumps to antimicrobial resistance is considered in detail in some comprehensive reviews (e.g., Menetrey et al., 2021; Chauviat et al., 2023).
It is noteworthy that efflux pumps SmeYZ, SmeDEF, and MacABCsm, besides their primary function of removing xenobiotics from bacterial cells, also impact motility, flagella formation, and biofilm development (Lin Y.-T. et al., 2014; Lin et al., 2015; Kim et al., 2018). Lin Y.-T. et al. (2014) demonstrated that a ΔsmeYZ mutant was unable to form flagella, resulting in a lack of motility, and exhibited reduced biofilm formation. The SmeYZ pump has been reported to contribute to a number of other physiological functions, including oxidative stress susceptibility, swimming, and, along with the SmeDEF pump, protease secretion (Lin et al., 2015; Wu et al., 2016; Kim et al., 2018). Blanco et al. (2019) reported that SmeGH is also involved in biofilm formation: a ΔsmeH mutant exhibited an elevated ability to produce a biofilm.
SmeYZ and SmeDEF are thought to be utilized by S. maltophilia against eukaryotes. Overexpression of SmeDEF in the S. maltophilia strain D457R led to reduced virulence against the social amoeba Dictyostelium discoideum (Alonso, 2004), and the loss of SmeYZ decreased in vivo virulence in a murine model and increased susceptibility to human serum and neutrophils (Lin et al., 2015). The above evidence significantly supports the suggestion that these RND pumps contribute to the S. maltophilia virulence.
The S. maltophilia MacABCsm differs from the MacAB homologs of other bacteria (Lin Y.-T. et al., 2014). In particular, the pump possesses its own cognate outer membrane protein (OMP), MacCsm; and the macABCsm operon is intrinsically expressed. Additionally, MacABCsm has a wider substrate range for extruding macrolides, aminoglycosides, and polymyxins compared to MacAB-TolC of E. coli (Lin Y.-T. et al., 2014).
Another noteworthy function of efflux pumps was described by Wu C.-J. et al. (2022). They revealed that the SmeYZ, SmeDEF, and SbiAB pumps along with other mechanisms, impact the secretion of the siderophore stenobactin and the utilization of iron ions (see below) (Wu C.-J. et al., 2022).
The SmeIJK efflux pump of S. maltophilia has been reported to be involved in cell envelope integrity and the envelope stress response. Huang et al. (2014) demonstrated that a smeIJK-deleted mutant has increased sensitivity to membrane-damaging agents (MDAs) compared to the wild-type strain and exhibited an increased RpoE-mediated envelope stress response. In addition, sublethal MDAs concentrations induced smeIJK expression in an RpoE-dependent manner.
Summarizing the above, the analysis of current data on efflux pumps suggests that the historically held belief regarding their main functions should be reevaluated, and bacterial efflux pumps are much more than antibiotic resistance determinants. Since pumps are revealed in both clinical and environmental strains (Youenou et al., 2015) and considering the environmental origin of S. maltophilia, the functions of efflux pumps may be linked to bacterial physiology and adaptation to various niches and environments, as well as coexistence within complex multi-species communities.
Virulence and ironIron is vital for the growth and proliferation of non-fermenting Gram-negative bacilli, including S. maltophilia. Competition for iron ions between bacteria and the host during chronic infections can be detrimental to the host. Bacterial iron uptake may lead to local tissue damage and systemic dysfunction, e.g., anemia of inflammation, also known as anemia of chronic disease, observed in infectious, inflammatory, autoimmune, neoplastic, and chronic kidney diseases (Jurado, 1997). Iron plays a role in bacterial pathogenicity and host defense mechanisms, which is often underestimated. In S. maltophilia, iron limitation induces biofilm formation, increases EPS production, and reduces the generation of reactive oxygen species (ROS) (Kalidasan et al., 2018). Therefore, bacterial systems aimed at acquiring and transferring iron ions into bacterial cells are considered significant VFs.
While a basic understanding of iron uptake in Gram-negative bacteria has been achieved, many molecular mechanisms involved in this process in S. maltophilia remain unclear. Similar to other bacteria, S. maltophilia possesses a number of iron acquisition mechanisms that exhibit functional redundancy (Figure 2).
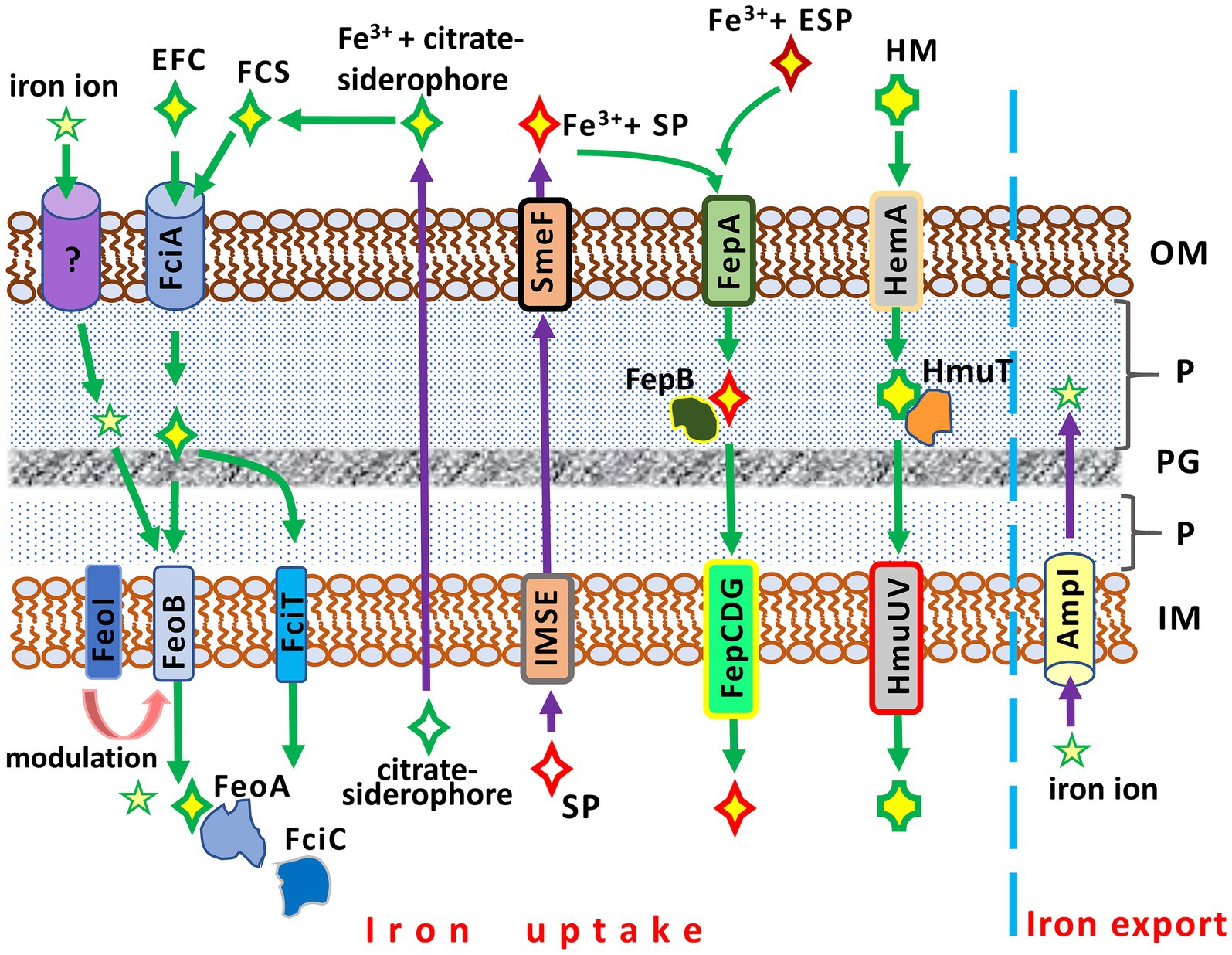
Figure 2. Iron acquisition and iron export systems in Stenotrophomonas maltophilia. IM, inner membrane; P, periplasm; PG; peptidoglycan; OM, outer membrane. AmpI, the inner membrane iron exporter; EFC, exogenous ferric citrate; ESP, an exogenous siderophore (siderophores produced by other bacteria, e.g., Pseudomonas aeruginosa); FciA, the outer membrane receptor for ferric citrate uptake; FciT (inner membrane protein) and FciC (cytoplasmic protein), putative proteins for citrate-mediated iron acquisition; FCS, the ferric citrate siderophore complex; FeoA, a cytoplasmic protein; FeoABI, the inner membrane transporter system of ferric citrate; FepA, the ferri-siderophore uptake system; FepB, a protein delivering ferric siderophore from the periplasm; FepCDG, the inner membrane transporter system for ferric siderophores; HemA, the TonB-dependent outer membrane receptor for hemin; HM, hemin; HmuT, a hemin-transporting protein; HmuUV, the inner membrane hemin transporter; IMSE, the inner membrane siderophore exporters (EntS for enterobactin, SmeY, SbiA, and SmeD for stenobactin); SmeF, the outer membrane exporter for siderophore; SP, S. maltophilia siderophore (stenobactin or enterobactin).
Two distinct iron uptake pathways are encoded by the S. maltophilia genome. These pathways include a siderophore- and a heme-mediated acquisition system (Kalidasan et al., 2018). The entAFDBEC operon controls the synthesis of the siderophore enterobactin, which belongs to the class of catecholamines, binding and transporting Fe3+ into the bacterial cell. The heme-mediated uptake system is under the control of the hgbBC and probably also the hmuRSTUV operons (Adamek et al., 2014; Kalidasan et al., 2018).
The simplest and least efficient absorption system is based on the diffusion of iron ions across OMPs (Liao et al., 2022). A more advanced iron uptake system employs siderophores, high-affinity iron-chelating molecules, to capture iron ions from the environment. These iron-siderophore complexes are recognized by specific OMPs and transported to the cytoplasm. S. maltophilia strains can produce at least two catecholate siderophores, stenobactin and an EntC-dependent catecholate siderophore which is sufficiently similar to, but distinct from, enterobactin of enteric bacteria (Nas and Cianciotto, 2017; Zhang et al., 2021; Wu C.-M. et al., 2022). Although one previous study (Jurkevitch et al., 1992) reported that S. maltophilia produces pseudobactin for iron uptake, this finding has not yet been confirmed. The export of siderophores is mediated by a system of membrane transport proteins (EntS, SmeY, SbiA, SmeD) of the inner membrane and TolC-exporter (SmeF for stenobactin) of the outer membrane (Wu C.-M. et al., 2022).
The ferric-enterobactin complex is recognized and taken up by the TonB-dependent FepA (ferric enterobactin protein), the enterobactin receptor on the outer membrane. Subsequently, FepB, the periplasmic binding protein, transports the iron-siderophore complex from the periplasm to the cytoplasm via the FepCDG transporter (Faraldo-Gómez and Sansom, 2003; Kalidasan et al., 2018).
Interestingly, while competing for iron, S. maltophilia can demonstrate cheating by using exogenous siderophores of xenogeneic origin. It has been reported that S. maltophilia can facilitate the uptake of ferri-pyochelin from P. aeruginosa in an iron-depleted condition (Pan et al., 2022).
An alternative to the catecholate siderophore is the citrate siderophore, which can be produced by S. maltophilia or obtained from exogenous sources. The Fe3+-citrate-siderophore complex is transported into the cytoplasm through a system of specific outer and inner membrane proteins (Figure 2). FciA (Sm
留言 (0)