Cerebral vascular pathologies including cerebral amyloid angiopathy (CAA) are a major contributor to the progression of neurodegenerative diseases, occurring in 80 % of Alzheimer’s Disease (AD) patients (Spina et al., 2021, Toledo et al., 2013). Despite its prevalence, CAA remains largely untreatable and is often overlooked (Sweeney et al., 2019). The hallmark clinical presentations of CAA include vascular cognitive impairment and dementia, recurring intracerebral hemorrhage and stroke (Reijmer et al., 2016). Vascular deposition of the amyloid-β (Aβ) peptide upon and within the cerebral vasculature is the pathological trigger of CAA. Although often considered concomitantly as both share Aβ deposition as a hallmark, AD and CAA are distinct diseases that can occur independently (Greenberg et al., 2020).
Aβ peptides originate from non-specific proteolytic cleavage of C99, the β-C-terminal fragment of the amyloid precursor protein (APP) (Wolfe et al., 1999). The cleavage event results in peptides of multiple lengths, with the major product being Aβ40 (Kummer and Heneka, 2014, Seino et al., 2021), the principal isoform in vascular deposits (Suzuki et al., 1994). The earliest familial form of CAA identified was linked to a Dutch family (Van Duinen et al., 1987) and originates from a glutamate to glutamine substitution at position 22 of the Aβ peptide (Van Broeckhoven et al., 1990). The resulting pathology, familial CAA Dutch (fCAA-Dutch), is an autosomal dominant form of vascular amyloidosis that is accompanied by accelerated cognitive impairment caused by extensive vascular amyloid deposition (Natté et al., 2001). fCAA-Dutch patients also spontaneously develop intracerebral hemorrhaging with the first stroke occurring on average at 25 years old (Van Duinen et al., 1987). Pathologically, the disorder is characterized by CAA type-2 Aβ deposition in cortical and leptomeningeal small arteries and arterioles with the absence of parenchymal cored amyloid plaques or neurofibrillary tangles that are key features of AD (Natté et al., 2001, Van Duinen et al., 1987).
The Aβ peptide is highly polymorphic, adopting a wide range of fibril conformations when incubated in solution (Tycko, 2014). However, it is unclear whether the structural variability observed in vitro reflects the presence of polymorphism in vivo (Cendrowska et al., 2020). The extent of structural variability of Aβ within a single patient remains debated. Some studies suggest one or two predominant structures exist per individual (Condello et al., 2018, Lu et al., 2013, Qiang et al., 2017), while others find variability within a patient (Rasmussen et al., 2017), or within amyloid plaques (Liu et al., 2016). Morphological differences between AD-specific parenchymal plaques and CAA-specific vascular deposits have been identified (Han et al., 2011, Rutgers et al., 2011, Schrag et al., 2011), suggesting a structural origin to the difference between the two pathologies. However, a robust link between structure and disease in the case of CAA has not been established, and the possibility of a structural origin to the variations in presentation, both between CAA subtypes and compared to AD, remains to be elucidated.
Recent studies have demonstrated structural differences between amyloidogenic proteins incubated in solution and those extracted from human patients (Schweighauser et al., 2020), including Aβ (Ghosh et al., 2021b, Kollmer et al., 2019, Wickramasinghe et al., 2021, Yang et al., 2022), suggesting that the cellular environment may influence fibril formation. The differences can arise in the shape or fold of the fibrils or in more subtle differences involving specific electrostatic or side chain packing interactions. For example, the in vitro structures of Aβ40 and Aβ42 reveal that electrostatic interactions of Lys28 may play a large role in fibril formation. In Aβ40, Lys28 interacts with Asp23, an interaction that allows the hydrophobic C-terminus to bend around and interact with the hydrophobic LVFF sequence. One interesting exception is the structure of the Osaka mutant (Schütz et al., 2015), which lacks E22. In this case, Lys28 interacts with Asp1 at the N-terminus of a neighboring protofilament within the fibril, yielding a very different fibril fold. In brain-derived Aβ40 fibrils, the D23-K28 interaction is generally preserved, but with differences in packing of the N-terminus or C-terminus within the fibril (see below). In Aβ42 with two extra C-terminal amino acids, Lys28 is oriented outward from the fibril center in in vitro fibrils and interacts with the C-terminal carboxyl group. This interaction induces a third β-strand in the overall fibril fold. In recent cryo-EM structures of brain-derived Aβ42 (Yang et al., 2022), the third β-strand is observed, Lys28 is oriented away from the fibril center, but the packing interactions within the fibril and between protofibrils are different than observed in in vitro structures.
Studies using electron cryo-microscopy (cryo-EM) (Ghosh et al., 2021a, Kollmer et al., 2019, Yang et al., 2022) and solid-state nuclear magnetic resonance (NMR) spectroscopy (Ghosh et al., 2021b, Lu et al., 2013) have investigated ex vivo Aβ fibril structures. Kollmer et al. (2019) (Kollmer et al., 2019) targeted vascular amyloid from cerebral meninges. They observed structural variability but were able to obtain a 4.4 Å structure of the predominant fibril population. The resulting structure revealed an ordered N-terminus and intermolecular hydrophobic packing, differing from structures of in vitro Aβ40 fibrils. The structure had some elements in common with fibrils seeded from parenchymal brain tissue using Aβ40 by Ghosh and colleagues (Ghosh et al., 2021a), although the latter exhibited a disordered N-terminal segment. It is not clear whether these structures are specific to the individuals studied or characteristic of their respective disease states.
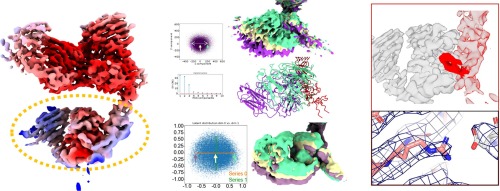
In this work, we compare the structures of Aβ40 fibrils seeded from vascular amyloid deposits isolated from sporadic CAA (sCAA) and fCAA-Dutch patients. We have recently shown that laser capture microdissection can be used to selectively isolate vascular amyloid deposits from brain slices (Irizarry et al., 2021). We use cryo-EM to determine fibril structures and identify structural elements unique to brain vascular-derived fibrils and use solid-state NMR spectroscopy to confirm the presence of these structural features. We find two structures prominently populate the patient-derived samples. Both structures exhibit a similar fold of the fibril core, characterized by an electrostatic cluster composed of residues D1-E22-D23-K28. Importantly, the highly ordered N-terminal segment composed of two β-strands in one population is unique to ex vivo fibrils. A structured N-terminus is generally not observed in fibrils formed in vitro. However, the N-terminus of C99 is folded into a two-stranded β-hairpin prior to cleavage by γ-secretase. We show here that K28 stabilizes the structured N-terminus in both the γ-secretase substrate and brain-derived fibrils, suggesting a role of the electrostatic cluster in APP proteolytic processing and in fibril formation in the human brain. The comparison of the fibril structures from sCAA and fCAA-Dutch patients highlights the importance of the D1-E22-D23-K28 electrostatic cluster in fibril formation and in disease progression in CAA and AD.


留言 (0)