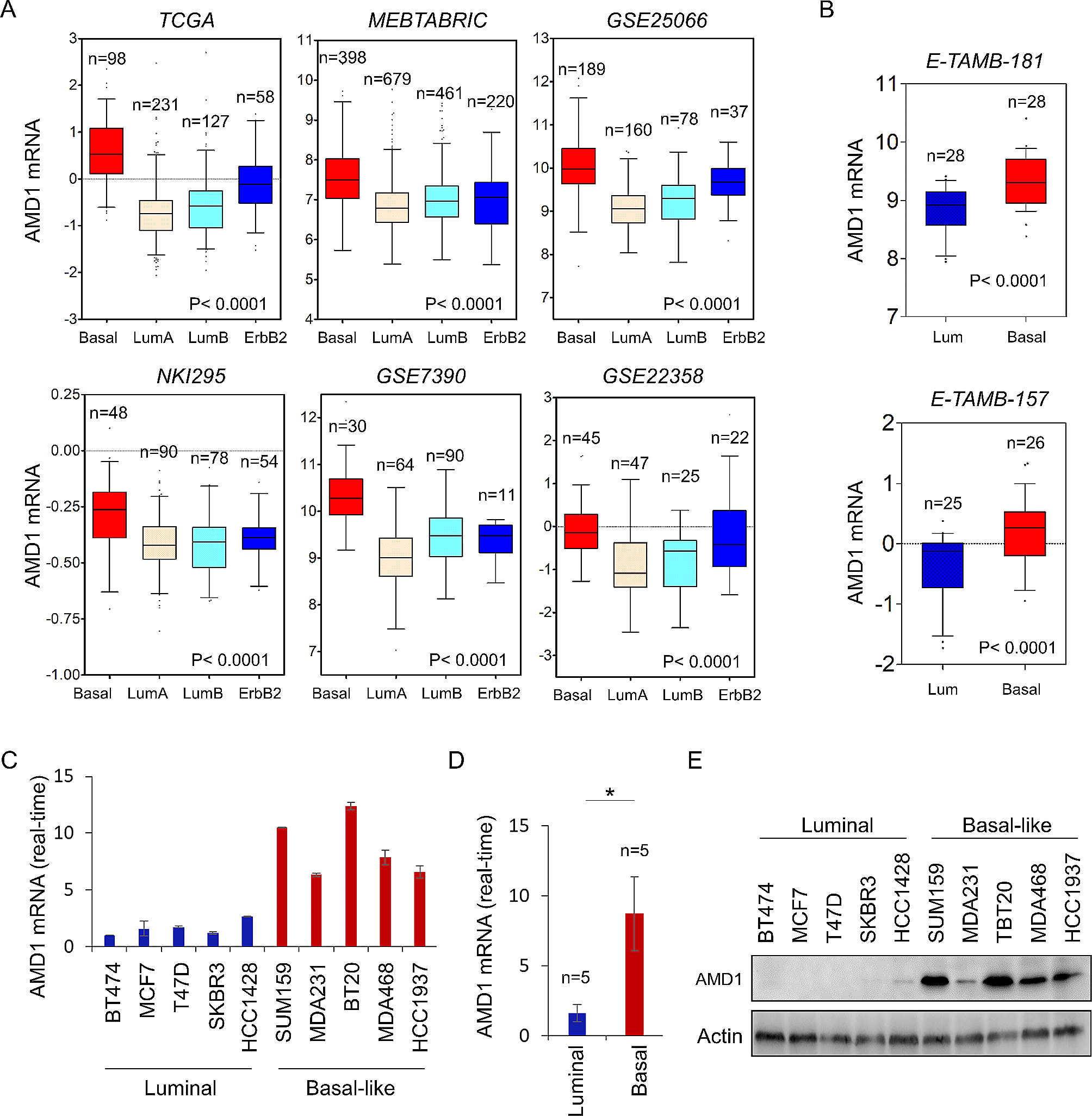
Patient population
HER2-amplified BrCa patients who underwent and successfully completed NAT with the TCbHP regimen and subsequent surgery at the Department of Breast Cancer Center, Chongqing University Cancer Hospital, from July 20, 2020, to May 28, 2021, were included in this prospective analysis. The study received approval from the Chongqing University Cancer Hospital’s ethics committee (CZLS2022022-A) and was carried out in strict conformity with the Good Clinical Practice guidelines and the Declaration of Helsinki. Informed consent form was signed by every patient.
The detailed other inclusion criteria were as follows: (1) histologically diagnosed with invasive breast carcinoma by core needle biopsy and subjected to IHC analysis with paraffin-embedded tumor samples biopsied before NAT. At least 1% immunoreactivity for either the estrogen receptor (ER) or progesterone receptor (PR) in tumor cell nuclei was required for a positive result. According to the 2018 American Society of Clinical Oncology (ASCO)/College of American Pathologists (CAP) Clinical Practice Guidelines, HER2 overexpression was indicated by a score of 3 + immunoreaction intensity or 2 + immunoreaction intensity with HER2 amplification by fluorescence in situ hybridization (FISH) [30]; (2) the lack of metastases, as determined by ultrasound, magnetic resonance imaging (MRI), computed tomography (CT), bone scan, and/or positron emission tomography (PET)/CT; When metastases to the axillary lymph nodes was either suspected or discovered, fine needle aspiration cytology was carried out; (3) having healthy kidney, liver, and hematopoietic systems as well as an echocardiography without significant cardiac arrhythmia or heart failure; (4) received the TCbHP regimen, which included carboplatin [area under curve (AUC) = 6], docetaxel [75 mg/m2, without dose escalation], a loading dose of trastuzumab (8 mg/kg) with a maintenance dose of 6 mg/kg, and a loading dose of pertuzumab (840 mg) with a maintenance dose of 420 mg every three weeks for six cycles.
The following conditions were excluded from the study: a history of prior malignancy (apart from inactive non-melanoma skin cancer and in situ cervical carcinoma), complicated with metabolic disorder syndrome, a current infection, and other concomitant illnesses that might impact medication tolerance or hamper compliance.
Evaluation of the NAT pathological response
Pathological response was gauged using the semi-quantitative Miller-Payne (MP) grading method. This gauges the percentage reduction in invasive tumor volume and cellularity following NAT based on the pathological evaluation of surgical samples [31]. In our investigation, pCR was determined by experienced pathologists to be the absence of residual invasive disease after surgery in both the breast (MP grade 5) and axillary lymph nodes (ypT0/is ypN0).
Sample collection
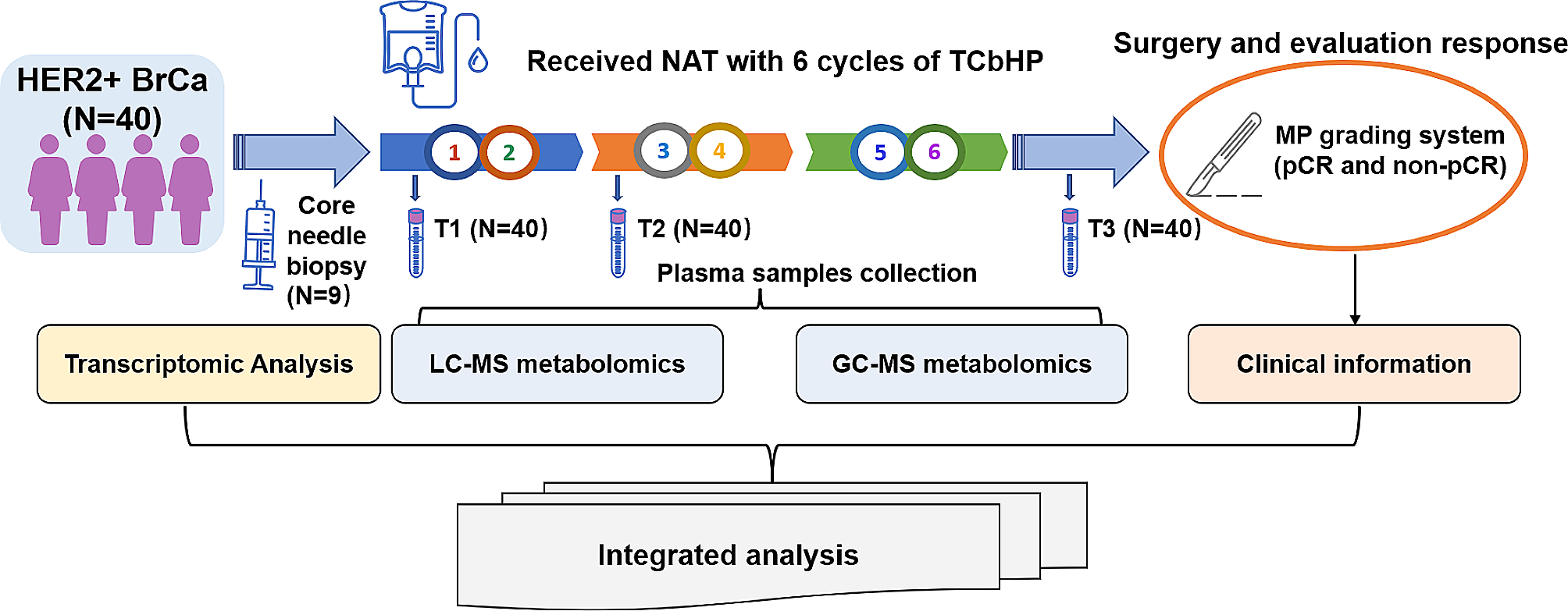
A total of 120 blood specimens from 40 patients were collected at three time points of NAT (baseline, T1; after 2 cycles, T2; after 6 cycles, before surgery, T3). Blood samples were taken from the elbow vein in the fasting state in the morning, kept in ethylenediaminetetraacetic acid vacuum tubes (BD Vacutainer, Franklin Lakes, NJ, USA), and centrifuged for 10 min at 3000 rpm at 4 °C. Immediately after separation, the serum was kept at -80 °C for further examination. A total of 1–2 cores of biopsies were taken from the breast tumor at the diagnosis and placed promptly frozen in liquid nitrogen before being stored at -80 °C until use.
Non-targeted metabolomic analysisGC-MS detection
The samples stored at -80 ℃ were thawed at room temperature. The following steps were taken to prepare the samples for GC-MS analysis: briefly, 450 µL methanol and acetonitrile (2/1, vol/vol) were used to extract the metabolites from the cecal content sample (150 µL). 50 µL BSTFA (with 1% TMCS) and 20 µL n-hexane were used to oxidate and derive the metabolites. During sample processing, ten different internal standards (C8/C9/C10/C12/C14/C16/C18/C20/C22/C24) were applied. Before the GC-MS analysis, the samples were left at room temperature for 30 min. Equal aliquots from each sample were combined to create the quality control (QC) sample.
The derivatized samples were examined using an Agilent 5977 A MSD system and an Agilent 7890B gas chromatography system (Agilent Technologies Inc., CA, USA). The derivatives were separated using a DB-5MS fused-silica capillary column (30 m × 0.25 mm × 0.25 μm, Agilent J&W Scientific, Folsom, CA, USA). As the carrier gas, helium (> 99.999%) was pumped through the column at a constant flow rate of 1 mL/min. The initial oven temperature was 60℃, held at 60 °C for 0.5 min, ramped to 125℃ at a rate of 8℃/min, to 210℃ at a rate of 5℃/min, to 270℃ at a rate of 10℃/min, to 305℃ at a rate of 20℃/min, and finally held at 305℃ for 5 min. The temperature of MS quadrupole and ion source (electron impact) was set to 150 and 230℃, respectively. The collision energy was 70 eV. Mass spectrometric data was acquired in a full-scan mode (m/z 50–500), and the solvent delay time was set to 5 min.
The QC sample was created by combining aliquots from each sample. Throughout the analytical run, the QCs were injected at regular intervals (every 10 samples) to generate a set of data from which repeatability could be evaluated.
LC-MS detection
At normal temperature, the samples frozen at -80 °C were defrosted. The following sample preparation procedures were used for LC-MS analysis: in brief, 150 µL of sample and 10 µL of L-2-chlorophenylalanine (0.06 mg/mL) dissolved in methanol were added to a 1.5 mL Eppendorf tube, and the tube was vortexed for 10 s. Then, 450 µL of an ice-cold mixture of acetonitrile and methanol (2/1, vol/vol) was added. The solutions were vortexed for 1 min, and the entire batch of samples was extracted using an ultrasonic device for 10 min in an ice-water bath before being kept at -20 °C for two hours. The extract was centrifuged for 10 min at 4 °C (13,000 rpm). A freezing concentration centrifugal drier was used to dry 150 µL of the supernatant in a glass vial. With the use of crystal syringes and 0.22 μm microfilters, the supernatants (150 µL) from each tube were collected and subsequently transferred to LC vials. Prior to LC-MS analysis, the vials were kept at -80 °C. A pooled sample made from an aliquot of each sample was used to create the QC samples.
To examine the metabolic profile in both ESI positive and ESI negative ion modes, a Dionex Ultimate 3000 RS UHPLC equipped with a heated electrospray ionization (ESI) source and a Q Exactive plus quadrupole-Orbitrap mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA) was used. In both positive and negative modes, an ACQUITY UPLC HSS T3 column (1.8 μm, 2.1 × 100 mm) (Waters Corporation Milford, Milford, MA, USA) was used. The binary gradient elution system consisted of (A) water (containing 0.1% formic acid, v/v) and (B) acetonitrile (containing 0.1% formic acid, v/v) and separation was achieved using the following gradient: 0.01 min, 5% B; 2 min, 5% B; 4 min, 30% B; 8 min, 50% B; 10 min, 80% B; 14 min, 100% B; 15 min, 100% B; 15.1 min, 5% and 16 min, 5%B. The flow rate was 0.35 mL/min and column temperature was 45℃. All the samples were kept at 10℃ during the analysis. The injection volume was 2 µL.
The mass range was from m/z 100 to 1,000. The resolution was set at 70,000 for the full MS scans and 17,500 for HCD MS/MS scans. The Collision energy was set at 10, 20 and 40 eV. The mass spectrometer operated as follows: spray voltage, 3800 V (+) and 3200 V (−); sheath gas flow rate, 35 arbitrary units; auxiliary gas flow rate, 8 arbitrary units; capillary temperature, 320 °C; Aux gas heater temperature, 350 °C; S-lens RF level, 50.
The metabolomic data processing
The plasma samples from patients in non-pCR (N = 19) and pCR groups (N = 21) at T1, T2, and T3 time points were termed as the A, B, C and D, E, F groups, respectively. The metabolomic analyses were based on these patients and groups.
To enable rapid data retrieval, the collected GC-MS raw data in.D format were converted to ABF format using Analysis Base File Converter program. The data were subsequently entered into the program MS-DIAL, which carries out peak detection, peak identification, characterization, MS2Dec deconvolution, peak alignment, peak filtering, and missing value interpolation. The LUG database (Untargeted metabolites database of GC-MS from Lumingbio) was used for the annotation of metabolites. A data matrix was derived. The sample information, the name of each substance’s peak, retention duration, retention index, mass-to-charge ratio, and signal intensity were included in the three-dimensional matrix. After screening, all peak signal intensities in each sample were segmented and normalized according to internal standards with relative standard deviation (RSD) greater than 0.3. Redundancy elimination and peak merging were carried out after the data had been standardized to produce the data matrix.
Progenesis QI V2.3 (Nonlinear, Dynamics, Newcastle, UK) was used to process the original LC-MS data for baseline filtering, peak identification, integral, retention time correction, peak alignment, and normalization. The main parameters used were 5% production threshold, 10 ppm product tolerance, and 5 ppm precursor tolerance. Using the Human Metabolome Database (HMDB) (https://hmdb.ca/), LIPID MAPS (V2.3) (https://lipidmaps.org/), Metlin (https://metlin.scripps.edu), EMDB, PMDB, and self-built databases to conduct qualitative analysis, compounds were identified based on the precise mass-to-charge ratio (M/z), secondary fragments, and isotopic distribution. The retrieved data were then subjected to additional processing, including removal of any peaks with missing values (ion intensity = 0) in more than 50% of the groups, replacement of zero values with half of the minimum values, and screening in accordance with the qualitative outcomes of the compound. Additionally, compounds that produced results of less than 36 (out of 60) points were declared incorrect and eliminated. The data from the positive and negative ions were integrated into a data matrix.
Then the matrix was imported into R (R package MixOmics) to perform unsupervised analysis of principal component analysis (PCA) to observe the general distribution among the samples and the stability of the entire analytic process. The metabolites that differed across groups were identified using supervised analysis of orthogonal partial least-squares discriminate analysis (OPLS-DA) (R package MetaboAnalystR), which were offen used to maximize the global metabolic variations among groups. Seven folds cross-validation and 200 response permutation testing (RPT) were employed to assess the model’s quality and minimize overfitting. The total contribution of each variable to group discrimination was ranked using the variable importance of projection (VIP) values derived from the OPLS-DA model. DEMs with VIP values higher than 1.0 and p-values lower than 0.05 were chosen. Hierarchical clustering analysis (pheatmap, R package pheatmap) based on these DEMs were utilized to demonstrate the expression pattern of DEMs in different groups and samples. The enriched pathway analysis of changed metabolites was performed using Kyoto Encyclopedia of Genes and Genomes (KEGG) database (http://www.genome.jp/KEGG/pathway.html) with the Hypergeometric Test to calculate significantly perturbed pathways. The pathway impact is the sum of the importance of the matched metabolites normalized by the sum of the importance of all the metabolites in each metabolic pathway.
Transcriptomic analysis
RNA isolation and high-throughput RNA sequencing (RNA-Seq) were performed by Oebiotech Corp (Shanghai, China). The Spin Column Bacterial Total RNA Purification Kit (Sangon Biotech, Shanghai, China) was used to extract the total RNA. RNA purity and quantification were evaluated using the Agilent 2100 bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). The NanoDrop2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, United States) was used to calculate the concentration. Integrity number (RIN) ≥ 7, 28 S/18S ≥ 0.7, and total RNA concentration greater than 0.5 µg were the selection criteria for RNA samples. For some of the enrolled patients had pathologic confirmation of BrCa when they consulted at our hospital or had insufficient residual tissue for transcriptomics test after pathology diagnosis, only 21 pre-treatment puncture specimens from these 40 patients were available for RNA extraction. After the quality control, only nine samples (43%), including six samples from patients in the non-pCR group (a03, a06, a12, a15, a17, a19) and three samples from patients in pCR group (d02, d08, d10), met the experimental requirements for RNA-Seq libraries construction; the other 11 samples had the RIN < 6–28 S/18S < 0.7, and one sample did not have sufficient total RNA for further experimentation. The libraries were constructed using VAHTS Universal V6 RNA-seq Library Prep Kit according to the manufacturer’s instructions. The transcriptome sequencing and analysis were conducted by OE Biotech Co., Ltd. (Shanghai, China).
The libraries were sequenced on a llumina Novaseq 6000 platform and 150 bp paired-end reads were generated. About 48.13 M raw reads for each sample were generated. Raw reads of fastq format were firstly processed using fastp and the low quality reads were removed to obtain the clean reads. Then about 6.7G CleanData for each sample were retained for subsequent analyses. The clean reads were mapped to the human reference genome GRCh38.p13 using HISAT2. Fragments per kilobase million (FPKM) of each gene was calculated and the read counts of each gene were obtained by HTSeq-count. Covariance-based PCA analysis were performed on the top 2000 highly variable genes with the highest degree of variation using R (v 3.2.0) to evaluate the biological duplication of samples.
Differential expression analysis was performed using the DESeq2 with negative binomial distribution (NB) test. Q value < 0.05 and foldchange ≥ 2 or foldchange ≤ 0.5 was set as the threshold for significantly DEGs. Hierarchical cluster analysis of DEGs was performed using R (v 3.2.0) to demonstrate the expression pattern of genes in different groups and samples.
Based on the hypergeometric distribution, Gene Ontology (GO, which provide annotation information of biological process, molecular function, and cellular component, http://www.Geneontology.org/) and KEGG pathway enrichment analysis of DEGs were performed to screen the significant enriched term using R (v 3.2.0), respectively.
Integrative analysis of metabolome and transcriptome
In this study, integrative analyses of gene expression differences (derived from nine patients including six patients from non-pCR group and three patients from pCR group) and metabolism differences (derived from 40 patients including 19 patients from non-pCR group and 21 patients from pCR group) between the non-pCR group and the pCR group at baseline were explored by transcriptomic analysis and untargeted metabolomic analysis. Based on the top 20 DEGs and DEMs, Spearman correlation coefficients were calculated by R and the cluster analysis heatmap was drawn. Then, all DEGs and DEMs were mapped to the KEGG pathway database, and common pathways information of them was acquired.
Cell lines and quantitative reverse transcription-polymerase chain reaction (qRT-PCR)
HER2 + BrCa cell lines, including trastuzumab-sensitive SK-BR-3 and BT-474 cells, primary trastuzumab-resistant JIMT-1 cells, and acquired trastuzumab-resistant SK-BR-3-HR cells (generated in our laboratory from the parental cell line SK-BR-3 by exposing the cells to gradually increasing concentrations of trastuzumab for 6 months), were cultured in appropriate medium. TRIzol solution (Thermo Fisher’s) was used to extract the total RNA from these cell lines, and a PrimeScript RT reagent kit (Yeasen, China) was used to reverse-transcribe the RNA into cDNA. qRT-PCR was performed using a LightCycler 480 (Roche) with a SYBR-based detection system, and specific primers were used to measure the relative mRNA expression levels of all the genes. The relative levels of the target genes to the control β-actin mRNA transcripts in each sample were analyzed by the 2-ΔCt method. Each experiment was run in triplicate. The primers utilized and their sequences are listed in the Supplementary Table 1.
Statistical analysis
Wilcoxon test was used for continuous variable (age at diagnosis), Chi-square test or Fisher exact test were used to compare dichotomous variables in other clinicopathological features analyses. The significance of metabolites in two groups or multiple groups was calculated by Wilcoxon test and Kruskal-Wallis test, respectively. The metabolites with VIP > 1.0 and P < 0.05 between two groups were considered as DEMs. RF was performed to screen potential metabolites for the prediction of treatment response to NAT (randomforest package) based on the pre-therapeutic DEMs between non-pCR patients and pCR patients. The two important parameters used in the RF classifier are ntree (number of trees) and mtry (the number of features to choose the best subset). The ntree parameter was set to 500 trees. The mtry was set to the default value (sqrt(p) where p is number of variables in the metabolomic data). The area under receiver operating characteristic (ROC) curve (AUC) (R package pROC) was applied to evaluate the performance of the predictive model based on the four pre-therapeutic metabolites selected by RF analysis. The correlation of the four pre-therapeutic metabolites with MP grade, and the correlation of DEMs and DEGs were calculated by Spearman correlation coefficients. The trends analyses of DEMs expression between non-pCR and pCR patients were performed by Mfuzz R package. The qRT-pCR results of several DEGs were presented as mean ± SD, and Student t test was used to generate P value between two cell lines. The visualization of results was performed by R (v 3.2.0) and GraphPad Prism software (v8.0). Unless otherwise stated, a variable was deemed to be statistically significant at P < 0.05.







留言 (0)