
記住我

Patients are recruited from a local hospital in Hong Kong. These patients will be followed up at the Orthopaedics Outpatient Clinic at the Prince of Wales Hospital for clinical examination and questionnaire filling. While for the muscle assessments, it will be conducted at the Sports Medicine and Rehabilitation Centre at the Chinese University of Hong Kong Medical Centre.
Eligibility criteriaThe inclusion criteria are as follows:
(1)Aged 18–50 with unilateral ACL injury
(2)Sporting injury with a Tegner score of 7 +
(3)Serum D level remained < 20 ng/ml
(4)Limb symmetry index of isokinetic quadriceps strength < 90% in the injured leg of the contralateral leg
(5)Both knees without a history of injury/prior surgery
The exclusion criteria are as follows:
(1)Concomitant bone fracture, major meniscus injury, or full-thickness chondral injuries requiring altered rehabilitation programme post-operatively
(2)Pre-operative radiographic signs of arthritis
(3)Metal implants that would cause interference on MRI
(4)Non-HS graft for ACLR
(5)Patient non-compliant with the rehabilitation programme
(6)Regular sunbed users
Who will take informed consent?Trained research assistants will obtain written informed consent from all participants prior to their participation in this study. Our research assistants will first explain to eligible participants our programme in detail. The study will be carried out in compliance with the Declaration of Helsinki and the ICH-GCP. Ethical approval will be obtained from the local IRB before the study starts. Informed consent must be obtained from all patients in order to participate in the study.
Additional consent provisions for collection and use of participant data and biological specimensConsent of participant data and biological specimens are also included in the informed consent. Blood specimens in this study will be disposed of after testing and will not be used for genetic analysis or used in other studies. Every participant will be represented by an ID number, and personal information such as name, address, and telephone number will be kept confidential before, during, and after the study.
InterventionsExplanation for the choice of comparatorsPatients randomized to the control arm will receive a placebo with the same appearance as vitamin D3. As a placebo will look and taste like vitamin D3, this can ensure the participants are blinded to the treatment. The utilization of a placebo that closely mimics the appearance and flavour of vitamin D3 will effectively blind the participants to their treatment.
Intervention descriptionFor the proposed study, we will use 2000 IU/day as advised by the Endocrine Society [10]. A previous study has shown that 2000 IU/day for 4 months showed significant improvement in muscle strength [22]. Therefore, for the proposed study, we will use 2000 IU/day for a duration of 16 weeks. Subjects will be randomized into two study groups: (1) placebo group—patients receive placebo; (2) intervention group—patients receive a daily dose of 2000 IU of vitamin D3 supplements. Supplements will be dispensed to participants in baseline and 1st follow-up to increase the subject compliance.
All study tablets including the supplement and placebo will be manufactured according to the Good Manufacturing Practice (GMP) guidelines for quality assurance. They will be taken with water at breakfast, one tablet at a time and once daily. The treatment will last for 16 weeks after which the supplementation will be stopped. Subjects will continue with their usual lifestyles in diet and physical activity without receiving any other treatment for muscle health.
Criteria for discontinuing or modifying allocated interventionsVitamin D has been considered safe for users when taken in appropriate doses [13]. Participants will be advised that in the event of nausea, vomiting, muscle weakness, or confusion, they should discontinue taking supplements and seek medical attention. The medical staff will perform the evaluation determining if immediate removal from the study is necessary for their best interest to safeguard their health.
Strategies to improve adherence to interventionsPatients will be contacted weekly for their intake of the intervention and 1 week before the assessment to enhance the attendance rate. Special assessment session on the weekend or in the evening will be arranged under special circumstances to enhance subject compliance. Patients who default a scheduled appointment will be contacted by the investigators to re-arrange another appointment within 1 week. In the case of patient non-compliance, study personnel would remove the patient from the study only when multiple failed attempts to contact and convince the patient occur. If the patient chooses to withdraw from the study before the end of the study period, the reason and termination date will be recorded. As far as possible, we will invite patients who would like to withdraw to attend the final assessment.
Relevant concomitant care permitted or prohibited during the trialParticipants remain on their standard treatment and medication procedures throughout the study period, and clinicians are advised to manage participants in the usual manner subject to the caveats outlined above.
Provisions for post‑trial careNot applicable, since ancillary and post-trial care is provided within the standard care.
OutcomesThe primary objectives of this study are to track the changes in peak torque and fatigue index (FI) of isokinetic muscle strength over a period of 6 months. The peak torque, measured in Newton metre (Nm), will be the highest recorded value among the 30 repetitions during the isokinetic muscle strength test, and the FI will be utilized to determine the per cent decrease for each variable that correlates with muscle endurance. Secondary outcome measures include (1) assessment of isometric muscle strength through quadriceps rate of torque development (RTD) and central activation ratio (CAR), (2) quadriceps muscle volume and muscle thickness, (3) evaluation of serum 25(OH)D using LC-Qtrap/MS, (4) measurement of passive knee laxity using KT-1000 knee ligament arthrometer (MEDmetric Corp., San Diego, CA, USA), (5) analysis on the reaming size of the bone using XtremeCt II, (6) the evaluation of ground reaction force will be carried out using a synchronized force plate located at the centre of the capture volume at 1000 Hz, (7) assessment of knee joint moments will be assessed by the skin marker-based motion analysis system, (8) distance by single-leg hop test, and (9) self-reported outcome assessing pain, disability, and activity level on knee function will be evaluated during the 6 months of follow-up.
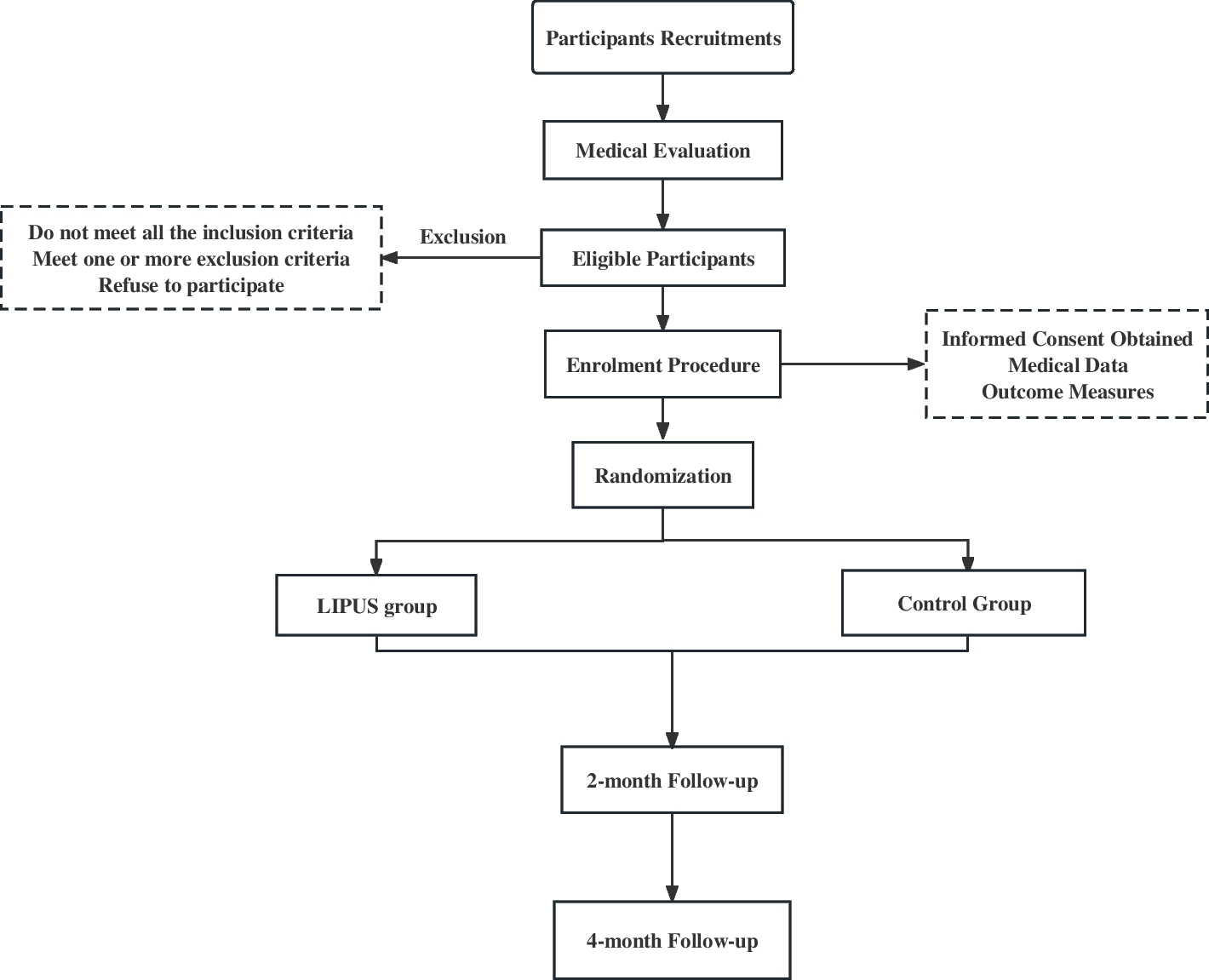
Participant timelineThe SPIRIT reporting guidelines were used to ensure the completion of the study protocol (Additional file 1) [4]. The study flowchart is illustrated in Fig. 1.
Fig. 1 Sample size
Sample size Quadriceps muscle strength will be employed as the primary outcome for sample size estimation. As reported [22], the difference in quadriceps between the two groups is 0.88. It is estimated that a sample size of 28 in each group will have 90% power to detect a significant difference using a two-sided independent t test with a 0.05 significance level (G*Power 3.1.9.4). Taking account of the 20% dropout rate, we further increase the sample size to n = 30 for each arm (total n = 60).
RecruitmentPatients who have persisting quadriceps muscle weakness after ACLR will be recruited consecutively.
The centre staff will assist in identifying eligible patients from the Department of Orthopaedics and Traumatology at Prince of Wales Hospital, Hong Kong, based on the inclusion and exclusion criteria and send them to us for screening. These patients will be screened at the Sports Medicine and Rehabilitation Centre at the Chinese University of Hong Kong Medical Centre. The patients will be explained the study procedures by the principal investigator. Patients who consent to participate in the study will attend a scheduled visit for baseline examination. The completion of the trial is expected to take 36 months.
Assignment of interventions: allocationSequence generation Randomization and blindingA total of 60 patients will be enrolled. Participants will be randomized into 1:1 allocation, blocked randomization with 30 participants in the vitamin D group and 30 participants in the placebo group. The randomization will be done using a computer randomization program before the intervention. This will be overseen by a biostatistician who is not involved in the recruitment of patients and data analysis. Hence, both participants and the research personnel are blinded until the completion of treatment.
Concealment mechanismAllocation concealment will be ensured as the computer randomization programme will not release the randomization code until the patient has been recruited into the trial. Vitamin D and placebo will be dispensed to participants in visually indistinguishable forms by Clinical Research Pharmacy based on the randomization code. Hence, patients and investigators are fully blinded until the completion of treatment. Outcome assessors and statisticians are also blinded.
ImplementationThe principal investigator will enrol participants. The computer randomization program will generate the allocation sequence at a 1:1 ratio. An independent research staff will assign participants to interventions.
Assignment of interventions: blindingWho will be blindedParticipants are blinded to the intervention. The research assistant who assists in consent seeking and monitoring of progress and adverse events will not be involved in outcome assessment. The assessor who will be another trained research assistant will be blinded to the randomization status and will not be involved in the intervention. The statistician responsible for the randomization is not involved in other parts of the study including data analysis.
Procedure for unblinding if neededIn normal circumstances, the blinding will be maintained unless a serious adverse event occurs. Unblinded participants will then exit the trial, and the medical conditions will be managed accordingly. The management results will be recorded on the clinical report form and reported to the Joint Clinical Research Ethics Committee of the Chinese University of Hong Kong and the New Territories East Cluster of the Hospital Authority.
Data collection and managementPlans for assessment and collection of outcomes Anthropometric measurementBody mass index (BMI) would be calculated by the measured height and weight. We would measure the waist circumference as well. Subcutaneous fat would be measured using skinfold techniques for the triceps brachii and biceps brachii.
Biochemical assaysBlood samples will be taken under non-fasting conditions. The serum obtained (5 ml) will be immediately stored at − 80 °C until analysis. Quantitative analysis for serum 25(OH) Vit-D assay will be performed using LC-Qtrap/MS.
Isokinetic muscle strength assessmentThe dynamometer (Biodex System 4, Biodex Medical Systems Inc., New York, USA) will be used. Prior to the test, the subjects will engage in a standardized warm-up exercise consisting of 5 min of cycling. The knee extension and flexion will be tested in concentric and concentric contractions at 60°/s and 180°/s [2]. Subjects will be seated on the dynamometer chair with their hips flexed to 85°. The speed of 180°/s is chosen to perform the fatigue test since high-speed workouts are expected to tire the fast-twitch muscle fibres more quickly [17]. For calculating peak torque, the single highest value within the 30 repetitions will be considered. The fatigue test will reflect muscle endurance, and the trends for peak torque, work, and power will be analysed. To calculate the percentage decrease for each variable, the FI (fatigue index) will be used [14]. The formula for percentage decrease is 100 − [(last 5 repetitions/first 5 repetitions) × 100]. If an individual fails to achieve their peak torque within the first 3 repetitions, a second F.T. will be calculated using the formula: Per cent decrease = [100 − [(last 5 repetitions/highest consecutive 5 repetitions) × 100]. The highest consecutive five repetitions will be determined by values attained from the repetitions immediately before and following the single highest repetition value [14]. The re-test reliability has been proven [18].
Isometric muscle strength assessmentAfter warming up on a stationary bicycle for 5 min, the strength of the quadriceps muscle will be measured using a Biodex dynamometer (Biodex System 4, Biodex Medical Systems Inc., New York, USA) through maximal voluntary isometric contractions (MVIC). The uninjured limb will be tested first, followed by the injured limb. To isolate knee movement, the participants will be stabilized with straps placed over the trunk, pelvis, and thigh, with the hip flexed at 90° and the knee flexed at 45°, respectively [11]. To get the participants accustomed and warmed up, three sub-maximal voluntary contractions will be performed. Afterward, the participants will be instructed to perform three 5-s MVICs, with a 30-s rest period between each contraction. During the contractions, participants will be motivated to “kick as fast and hard as possible” verbally. The highest peak torque achieved among the three contractions will be collected as the MVIC and normalized by body mass for analysis.
The quadriceps rate of torque development (RTD) will be obtained from the MVIC test. The early and late RTD values will be calculated by determining the average slope of torque versus time curve measured from 0 to 50 ms (RTD0-50) and 100 to 200 ms (RTD100-200), respectively, after the onset of MVIC [8]. The onset of a contraction is identified as the torque ≥ 20 Nm. The highest RTD will be selected and normalized to body mass for analysis.
The superimposed burst technique (SIB) will be used to measure activation failure. This technique delivers a series of electrical stimulations to the quadriceps during a MVIC, causing a transient increase in muscle torque. All participants will be given a minimum of 5 min of rest after completing the MVIC test to prevent muscle fatigue. The position of SIB is the same as the MVIC test on the Biodex dynamometer. Two self-adhesive electrodes [ValuTrode (7.5 × 13 cm), Axelgaard manufacturing, CA, USA] will be attached to the quadriceps along the femoral nerve. All participants will be instructed to perform three additional 5-s quadriceps MVICs. A 1-min rest interval is provided between each contraction. During the MVIC of the quadriceps, an electrical stimulator (DS7R; Digitimer, Welwyn Garden City, UK) controlled by the Signal 7.05a software (CED Software, Cambridge, UK) will automatically deliver a supramaximal electrical stimulus. The stimulus consisted of 10 pulses with a pulse duration of 600 μs, delivered at a rate of 100 pulses per second. This occurred at the 3rd second of the MVIC. The intensity of the supramaximal electrical stimulus is determined prior to the SIB test. Electrical stimulations will be progressively delivered to the quadriceps, increasing by 100 mA each time until the stimulation-induced torque reached a plateau at rest. To ensure full stimulation of the quadriceps, the SIB test utilizes a current intensity of 120% plateau. In order to ensure maximal participant exertion, a successful trial is defined as achieving greater than 90% MVIC of quadriceps torque prior to the electrical stimulation. To represent full activation of the quadriceps, a central activation ratio (CAR) will be utilized. The following formula is used to calculate CAR: CAR = MVIC/(MVIC + stimulation provoked torque (SPT)). For analysis, the highest CAR obtained from three successful trials was recorded.
Radiological assessmentUltrasound imaging: The muscle thickness of the vastus medialis (VM), vastus lateralis (VL), and rectus femoris (RF) on both the injured and uninjured leg will be measured using the Aixplorer® ultrasound system (SuperSonic Imagine, Aix-en-Provence, France) and a linear transducer probe with a bandwidth of 2–10 MHz (SuperLinear™ SL10-2, Vermon, Tours, France). The participants will lie on a treatment table in a supine position during the assessment. A measuring tape will be used to locate VM, VL, RF, and the patella by palpation, and then marked with a pen for reference. Following the guidelines below, we consistently measure and label the locations as the three muscle groups for ease of comparison across patients. The following are the locations of specific points on the leg: RF is located at half of the distance from the anterior superior iliac spine (ASIS) to the superior pole of the patella, VM is located at one-fifth of the distance away from the midpoint of the medial patella border to the ASIS, and VL is located at one-third of the distance from the midpoint of the lateral patella border to the ASIS. After locating the anatomical points, excess contact gel will be applied to these points. The transducer probe will be aligned in the transverse plane and moved along the entire muscle bundle to capture a view of the VM, VL and RF. The operator will position the probe into the sagittal plane to measure muscle thickness upon the marked anatomical points. Minimal pressure will be applied to the limb to prevent muscle deformation. The results will be derived from three measurements averaged.
HR-pQCT: The HR-pQCT (ExtremCT II, Scanco, Switzerland) will be used to measure the size of the bone shell at the graft tunnel interface in the injured knees. The scan will consist of a total of 1344 axial slices with an image matrix of 2304 × 2304, taken at a nominal isotropic voxel size of 60.7 μm. The scan region will be defined by the scout view image that is acquired in the sagittal plane. The total scanning length will be 81.6 mm, covering the tunnel from the proximal tibia to the distal femoral condyle. The X-ray settings used will be 68 kVp, 1470 μA, 100 ms integration time, and 156 mAs per stack (168 slices). To avoid artefacts, patients will be instructed to sit still during the measurement. The image segmentation will be performed to select the bone shell features at graft tunnel interfaces near the femoral and tibial intra-articular apertures. This will be done using standardized threshold values at a thickness of approximately 2.5 mm (40 slices). The geometric transformation will be used to adjust for variations in the angle between the scanning axis and the tunnel axis by using a cosine function. The bone shell size will be presented as a summation of the segmented angle of incidence (AOI) of 40 slices to yield a volume of interest (VOI) in μm3.
Passive knee laxityTo measure the anterior–posterior knee laxity, the KT-1000 knee ligament arthrometer (MEDmetric Corp, San Diego, CA, USA) will be used. A manual force test will be applied until a 30-lb sound signal is activated. Three trials will be performed.
Biomechanics-motion analysisThe lower-body marker set-up will be used to assess kinematics via a skin marker-based motion analysis system (Vicon MX, Oxford, UK) following the OSTRC standard protocol, utilizing 16 cameras and 16 reflective skin markers. The kinetic variables including vertical and horizontal ground reaction force (GRF) and joint moments will be measured using a synchronized force plate (0.60 × 0.40 m, model OR6-7, AMTI, Watertown, MA) at the centre of the capture volume at 1000 Hz.
Single-leg hop (SLH) task: The SLH test will be performed as reported in the previous studies [19]. Three trials will be performed on each leg followed by familiarization. The SLH test will be considered valid if the patients can hop the maximum distance while keeping their balance for at least 2 s after landing.
Single leg squat (SLS) task: The subjects will begin by standing upright with their toes pointed forward and then squat down at their own pace. Once they reach the designated flexion angle, they will be asked to hold the position for ten seconds. If the subject is unable to maintain their balance, the trial will be deemed invalid. All participants will practice enough to achieve the required knee angle, which is between 40 and 50 degrees.
Self-reported outcomesLysholm Score: Lysholm Knee Score is a questionnaire that examines knee-specific symptoms and function of daily living. It consists of eight items, with a total score ranging from 0 to 100 and a higher score indicates a better outcome with fewer symptoms of disability.
International Knee Documentation Committee Subjective Knee Form (IKDC): IKDC is a self-reported questionnaire that measures symptoms, knee function and activity of daily living. The questionnaire consists of 10 questions, with a total score ranging from 0 to 100 and a higher score indicates greater knee function.
Tegner Score: The Tegner Activity Scale will be used to assess activity levels related to sports on a scale of 0 to 10. Zero represents a low activity level, and 10 represents the highest activity level.
Physical Activity Questionnaire: The level of physical activities during the past year will be evaluated with a validated Chinese version of the quantitative physical activity questionnaire adapted from Baecke et al. [9].
Food Frequency Questionnaire: The level of estimated vitamin D level taken from food will be evaluated with a validated Chinese version of the food frequency questionnaire.
Sunlight Exposure Questionnaire: The level of estimated vitamin D level absorbed from sunlight exposure will be evaluated with a validated Chinese version of the sunlight exposure questionnaire.
The assessment schedule is shown in Table 1.
Table 1 Assessment schedulePlans to promote participant retention and complete follow-upPatients will be contacted weekly for their intake of the intervention and 1 week before the assessment to enhance the attendance rate. Special assessment session on the weekend or in the evening will be arranged under special circumstances to enhance subject compliance. Patients who default a scheduled appointment will be contacted by the investigators to re-arrange another appointment within 1 week.
Data managementClinical data will be collected and recorded by trained research assistants in our research centre. Clinical examination data will be entered on case report forms and then entered electronically. Consistency checks by another technician will be performed to ensure data entry accuracy. All data will be stored in password-protected computers. The study will be conducted in compliance with Good Clinical Practices to ensure the rights and well-being of the participants and that the data collected are complete and verifiable from source documents. Patients are free to withdraw from the study at any time without giving any reasons, and their medical care or legal rights will not be affected. Patient files will be maintained in storage for a period of 3 years after completion of the study.
ConfidentialityAll personal information and consents on enrolled patients collected on paper versions will be kept in locked units at the participating practice and later at the coordinating centre to be archived. Each patient will be assigned an identification code. All information collected and inputted on the electronic database will be based on the identification code and therefore does not contain any personalized information that enables the identification of the patient. The document containing the information on the identification code and the identity of the patient will be kept separate from the study data files and data sheets. The patient identification code list and database can only be retrieved by dedicated study team members or be inspected by study monitors for quality checking and verification.
Plans for collection, laboratory evaluation, and storage of biological specimens for genetic or molecular analysis in this trial/future useBlood will be collected at the clinical sites for evaluation. After arrival at the local research laboratory at each site, the samples will be collected, transported, stored, and prepared according to local protocols. Blood will be stored at 2–8 °C before handling within the required time. All samples collected during the trial will be labelled with the patients’ identification code and will not contain any identifiable data. Patients have the option of consenting to their samples being stored for future research uses. Samples will be stored anonymously at a central location for a minimum of 5 years and a maximum of 10 years after completion of the study, after which these specimens will be destroyed by incineration according to local guidelines and protocols. The potential usage of the stored samples is included in the informed consent form. Nevertheless, further usage of the samples will need to be approved by the institutional ethical committee.
留言 (0)