
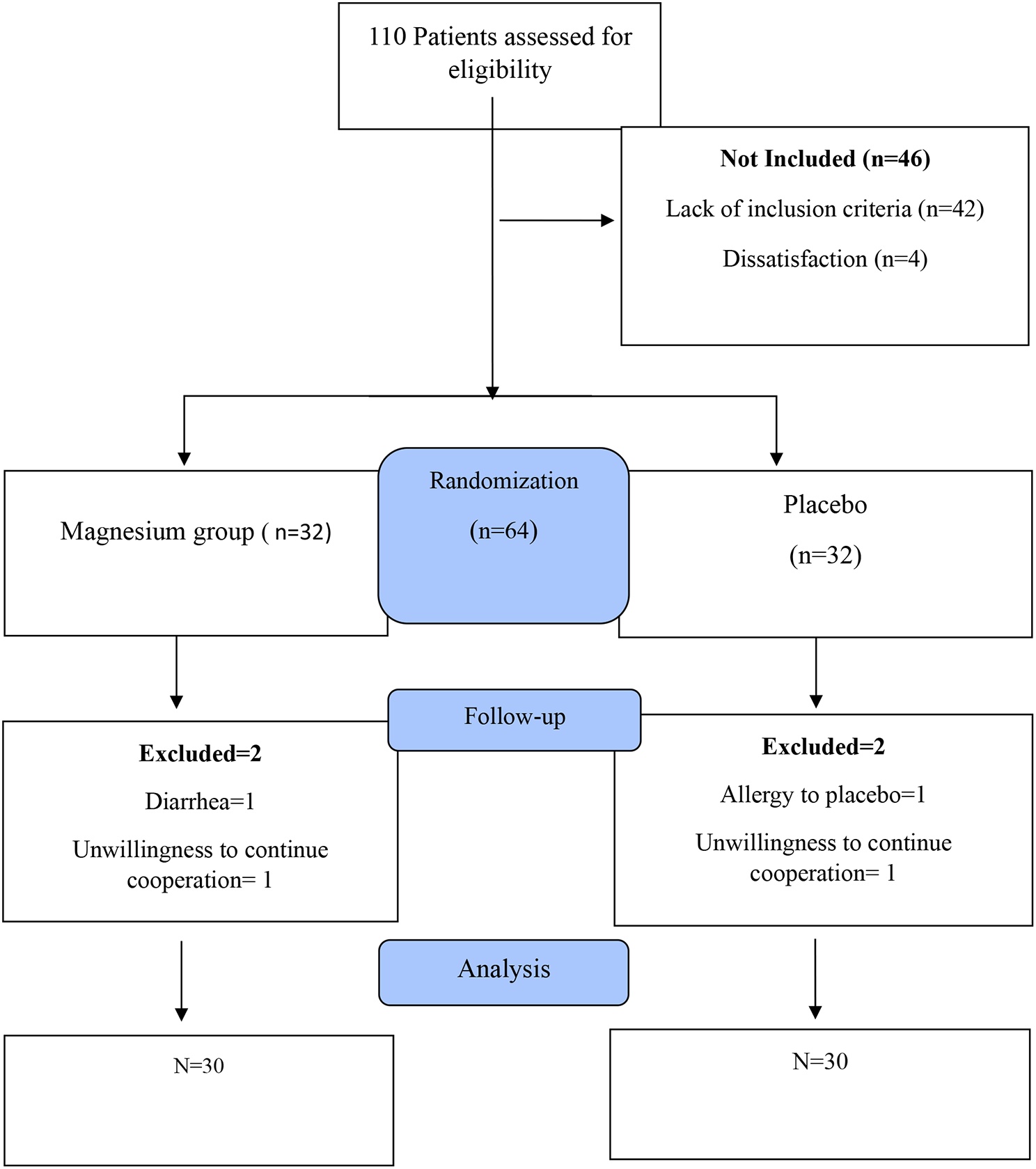
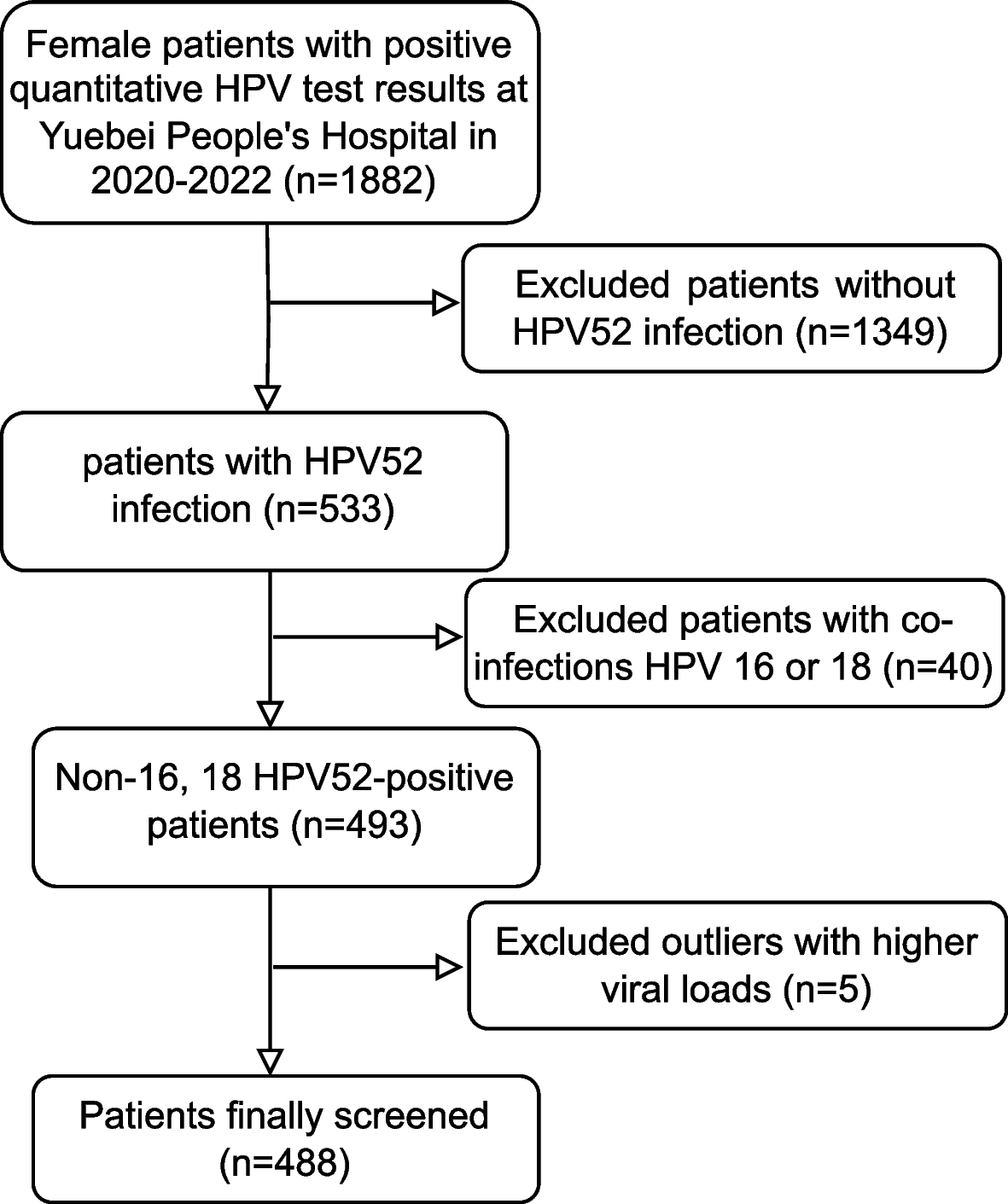
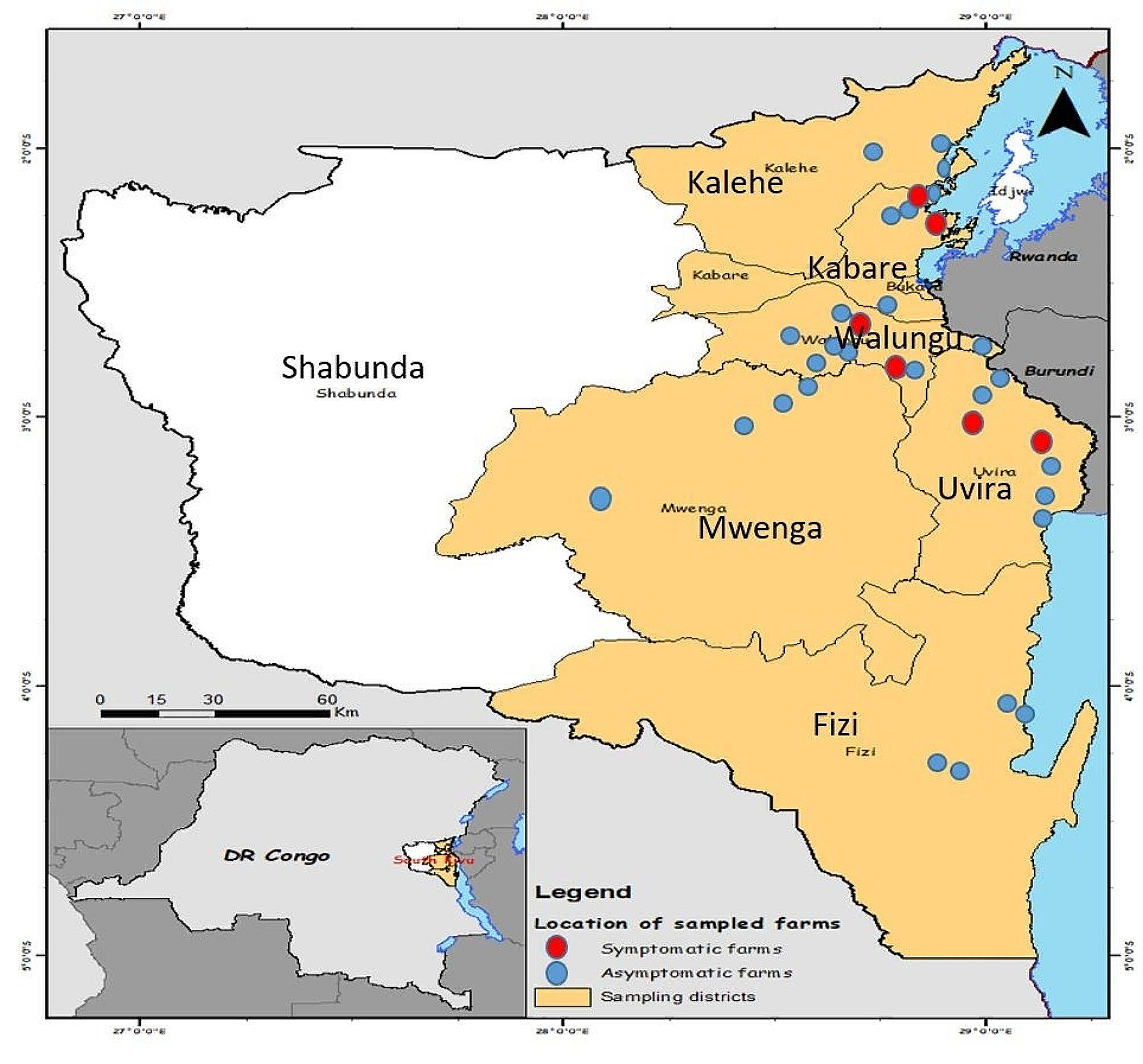
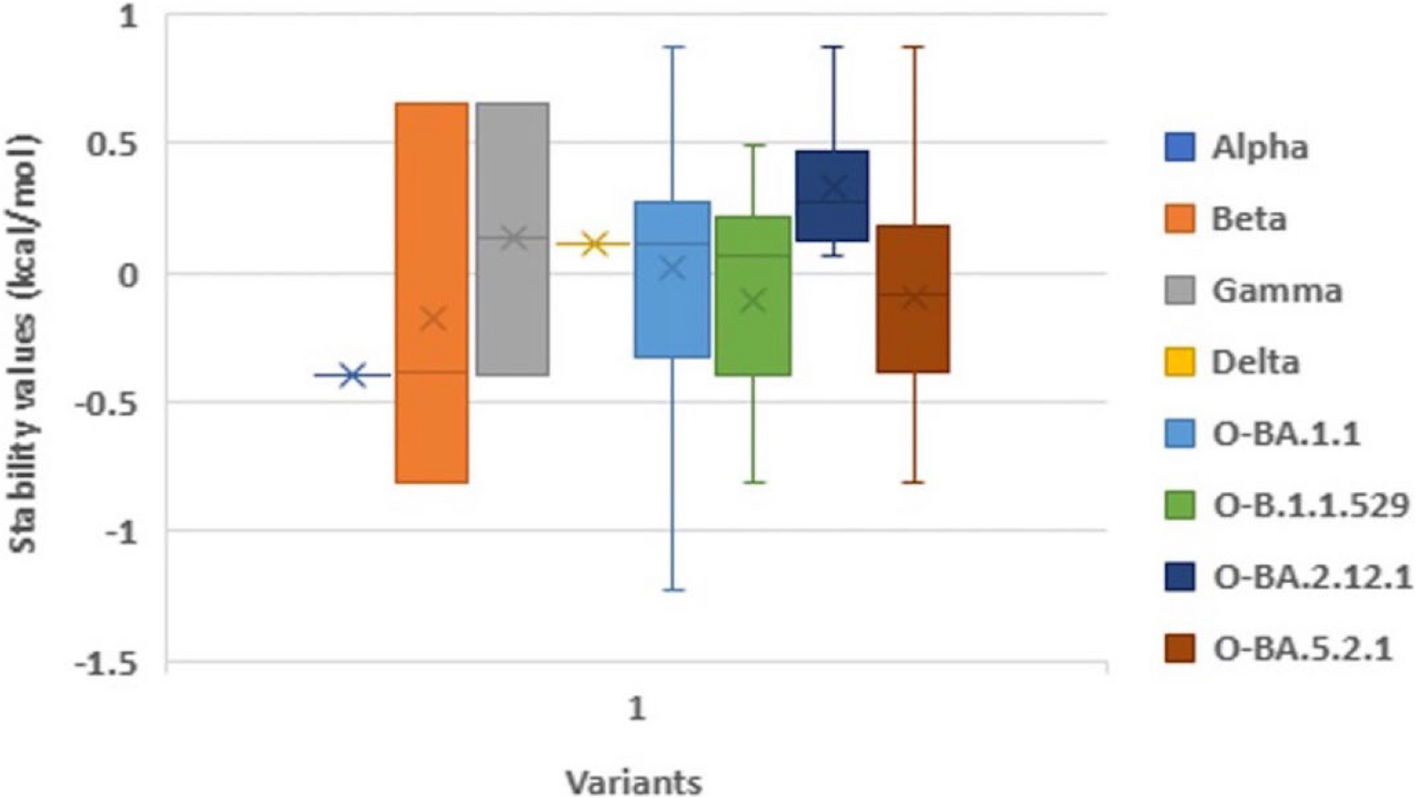
記住我
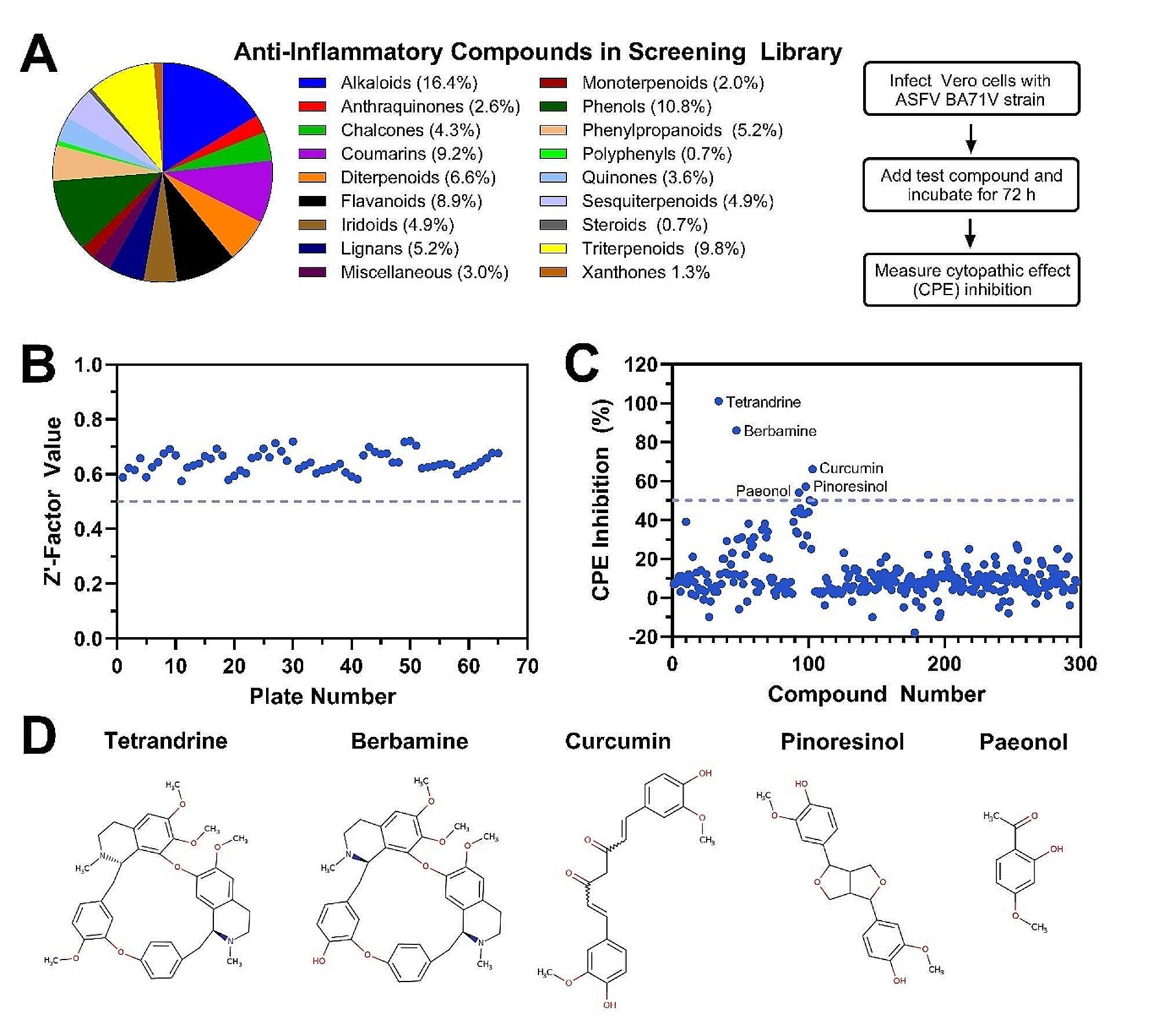

In this study, a total of 934 samples were collected. Nucleic acid testing revealed that the positive detection rate of influenza A virus in the LPM was 43.66% (289/662), in poultry processing plants was 29.33% (44/150). No positive samples were found on extensive poultry farms (0/120).
Among the positive samples (n = 333), nucleic acid typing revealed a detection rate of 70.87% (236/333) for the H9 subtype, 27.03% (90/333) for the H5 subtype, and 16.82% (56/333) for the untyped. No H7 subtype was detected in any of the samples.
Among the samples with Ct values less than 30 (n = 130), 119 sequencing results were obtained. Of these, 63 had single results (for which only one virus was detected), and 56 had mixed results (for which multiple viruses were detected simultaneously). The AIV subtypes detected included H1, H2, H3, H4, H5, H9, and H11, with detection rates of 0.84% (1/119), 1.68% (2/119), 14.29% (17/119), 5.88% (7/119), 72.27% (86/119), 60.50% (72/119), and 0.84% (1/119), respectively. Amino acid sequence analysis of the HA protein cleavage site revealed that a total of 37 samples were non HPAIV samples.
Virus isolation and resequencingWe successfully isolated 27 AIVs and determined their complete genome sequences through resequencing. The virus isolation rate was 72.97% (27/37), and the resequencing completion rate was 100% (27/27). Of the 27 resequenced virus strains, 23 were identified as H9N2, one as H3N2, one as H3N8, one as H4N8, and one as H4N1, with the latter being notably rare.According to the established nomenclature for influenza viruses, the strain of H4N1 subtype AIV was named A/environment/chongqing/cs2301/2022 (abbreviated as cs2301) and deposited in the GISAID database under the accession number EPI_ISL_18455199.
Gene analysisWe conducted a comprehensive search of the GISAID database and identified a total of 43 H4N1 virus sequences. This data volume was very small. After meticulous removal of duplicate or incomplete data, we obtained a set of 15 nonduplicate and complete sequences. Subsequently, we compared the gene homology between this set of sequences and cs2301. (Fig. 1).
Fig. 1
Comparison of gene homology between cs2301 and other H4N1 strains. The X-axis represents the gene fragments, while the Y-axis represents the isolate IDs. Different colors on the graph represent different levels of similarity expressed as a percentage
Our analysis revealed that, among the other H4N1 sequences, cs2301 exhibited the highest similarity (> 90%) with the EPI_ISL_697696 sequence. Furthermore, the internal genes of cs2301 exhibited a high degree of similarity (> 90%) with the EPI_ISL_130351 sequence.Compared to other H4N1 strains, these three strains share a closer genetic relationship.
We compiled essential information on H4N1 and conducted an analysis of key sites in the HA gene. Additionally, we predicted potential glycosylation sites on the HA protein (Table 1).
Table 1 Basic information, HA cleavage region, receptor-binding pocket and HA glycosylation site of H4N1Through the analysis of basic information such as location, collection date, and host species related to the H4N1 subtype, we revealed that the H4N1 subtype is distributed across regions including North America, Eurasia, and Australia. The first isolation of H4N1 was in North America in 1976, followed by subsequent isolations reported in Europe, Asia, and Australia from 1998 to 2022. These data suggest a broad temporal and geographic distribution of the H4N1 subtype. The primary hosts of H4N1 included multiple species of wild waterfowl (9/14) and domestic waterfowl (4/14), with a single case of H4N1 infection in a pig reported in China in 2009 [13]. This diversity of hosts suggests a broad host range for the H4N1 subtype.
Through the analysis of key sites in the HA, we revealed that H4N1 lacked consecutive basic amino acids at the HA cleavage site, which classifies them as LPAIV. Furthermore, their receptor-binding pocket sequences and glycosylation sites show a high degree of similarity.
Phylogenetic analysisWe performed a BLAST analysis of the eight genes of the cs2301 strain against the GISAID database, resulting in the identification of 250 sequences with the highest similarity. Phylogenetic trees were constructed for each gene after removing similar sequences (Fig. 2).
Fig. 2
Phylogenetic tree of HA (A), NA (B), PB2 (C), PB1 (D), PA (E), NP (F), MP (G) and NS (H). The sequences were aligned using the Muscle method. A phylogenetic tree was constructed using the neighbor-joining method, and the bootstrap values were determined through 1000 replications.The bootstrap value is represented by solid circles on the branching nodes. Different colors of the text represent different hosts, while different colors of the branches indicate different geographical locations of separation. The scale bar represents the distance unit between sequence pairs
Among the HA genes, the virus hosts exhibiting the highest similarity to cs2301 were primarily wild waterfowl (72/146) and domestic waterfowl (54/146). The regions with the highest number of isolates were Northeast Asia (37/146), North Asia (28/146), and East China (21/146). cs2301 was shown to share close phylogenetic ties with H4N2 subtype AIVs isolated from hosts such as Mallard and Mandarin ducks in Shanghai, East China region in 2019 (bootstrap values = 83).
Regarding the NA gene, the virus hosts with the highest similarity to cs2301 were primarily wild waterfowl (88/151) and domestic waterfowl (35/151). The regions with the highest number of isolates were Northeast Asia (47/151) and North China (24/151). cs2301 was closely related to H6N1 subtype AIVs isolated from wild waterfowl, such as Mallard and Anser_brachyrhynchus, in Anhui, Central China, and Korea in Northeast Asia in 2019 (bootstrap values = 96).
Among the internal genes of cs2301, the PB2 gene exhibited a close relationship with A/Common Teal/Shanghai/NH21680/2021 (H4N2), the PB1 gene with A/Common Teal/Shanghai/NH21920/2021 (H4N2), the PA gene with A/Environment/Chongqing/01452/2021 (H3N2), the MP gene with A/duck/Bangladesh/17D1589/2021 (H6N1), and the NS gene with A/Environment/Chongqing/01458/2021 (H3N2) and A/Environment/Hunan/13,561/2020 (H3N2). The bootstrap values supporting these relationships were greater than 70. However, the NP gene of cs2301 could not be clustered with the closest group of influenza virus genes in the database.
Mutation locus analysisWe conducted FluSurver analysis of the HA, NA, PB2, and MP genes of cs2301. According to the evolutionary analysis results, A/Duck/HongKong/24/1976 (H4N2) was chosen as the reference for HA and PB2, while A/Duck/Taiwan/0526/1972 (H6N1) was selected as the reference for NA and MP. A total of 32 notable mutation sites were identified. (Table 2).
Table 2 Notable mutation sites in cs2301Compared to the reference sequence, cs2301 exhibited 7 mutations in the HA gene, 2 in the PB2 gene, 18 in the NA gene, and 3 in the MP gene. These mutations have the potential to affect the structural interactions of the virus. Notably, certain mutations, such as T140S and E173A in the HA gene, as well as N222D and S248N in the NA gene, have multiple positional effects, encompassing host specificity shift, antigenic drift, immune escape, and mild drug resistance. Additionally, the E173A mutation in the HA gene and the I292V mutation in the PB2 gene have previously been reported to be associated with host-specific shift.
留言 (0)