
記住我
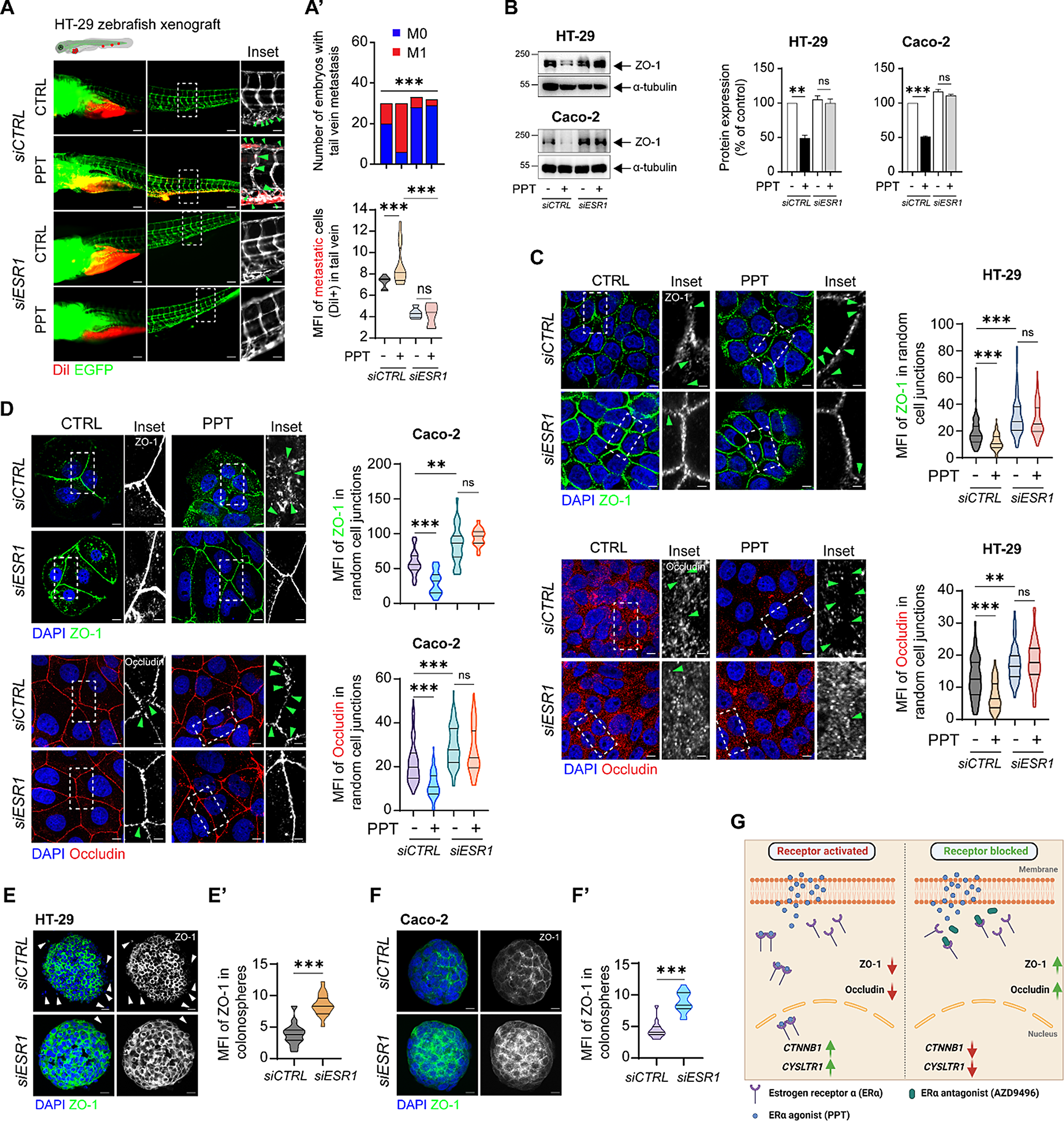

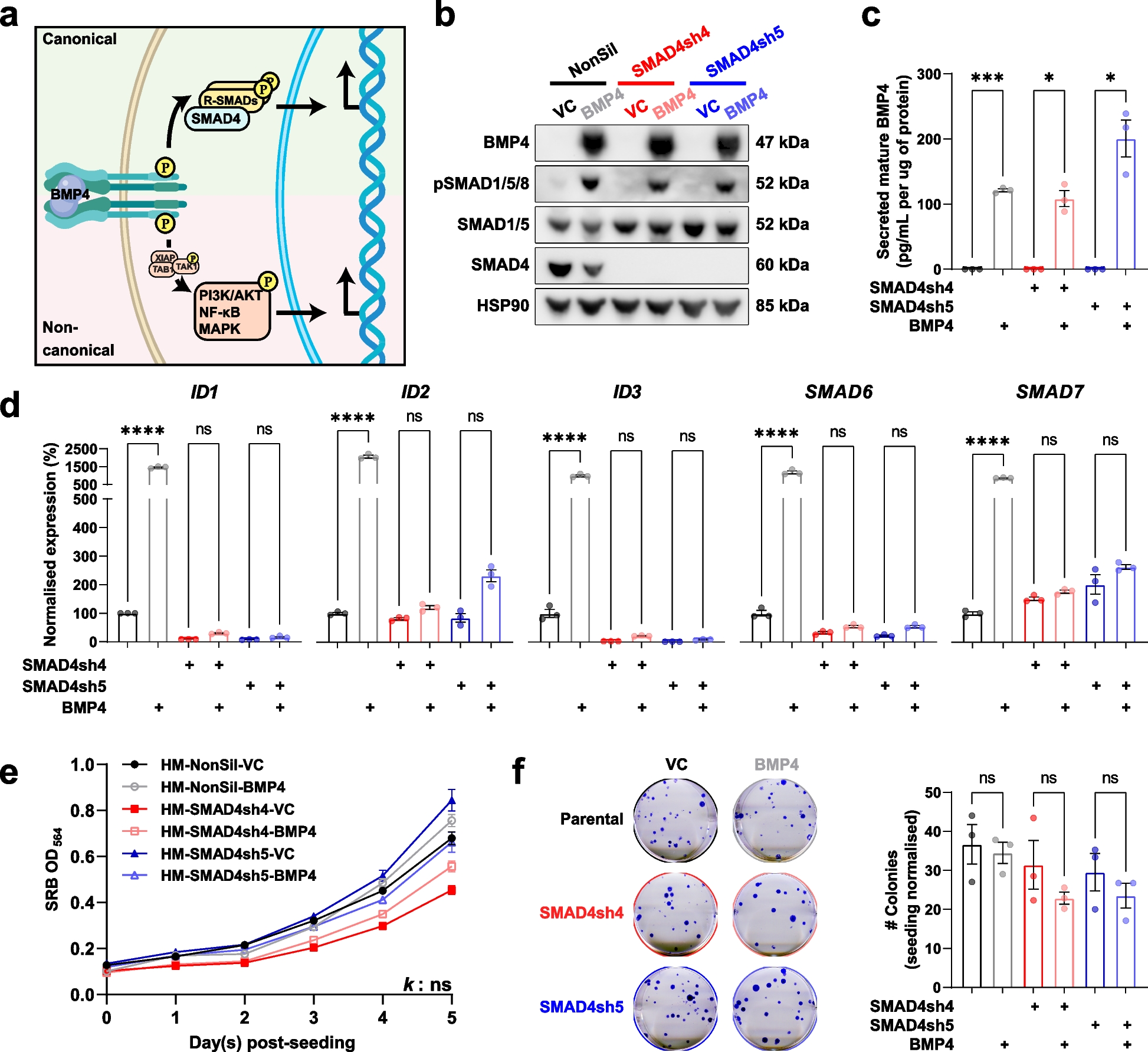


Although the AC-interacting domains of Gαi1 have not yet been elucidated, designing an effector-deficient Gαi1 chimera to test for abolishment of QL/RC-driven constitutive activities was made feasible by previous mutagenesis and structural studies of other Gα subunits (such as Gαi2), because Gαi1-3 show remarkably high homology (~ 90% with respect to Gαi1) [29]. Moreover, several regions identified in previous mapping studies [17,18,19, 27, 30] correspond to potential effector binding sites in the crystal structures of Gαt1 and Gαs [20, 31]. These domains include the switch II region, switch III region, α3 helix, αG/α4 loop, α4 helix and the α4/β6 loop, and molecular modeling of Gαi1 revealed that they may provide a planar surface for protein–protein interaction (Fig. 1A). It is likely that Gαi1 employs one or more of these regions to interact with AC.
Fig. 1
Putative AC-interacting domains of Gαi1. A The 3-dimensional structures of the GTPase domains of inactive (gray, PDB code: 1GP2) and active Gαi1 (yellow, PDB code: 1GFI) are overlaid and displayed as side, top and expanded views. The putative AC-interacting domains are marked in pale green (for side and top views) or labeled in the expanded view. Residues that are strictly conserved in AC-inhibiting Gαi1-3 and Gαz are shown as cyan (inactive) or orange (active) sticks in the expanded view. B Amino acid sequence alignment of the putative AC-inhibiting regions between Gαi1-3, Gαz, and the homologous regions of Gαt1 and Gαq. Conserved residues are indicated in orange. Residues subjected to point mutations in the chimeric studies are annotated with green dots
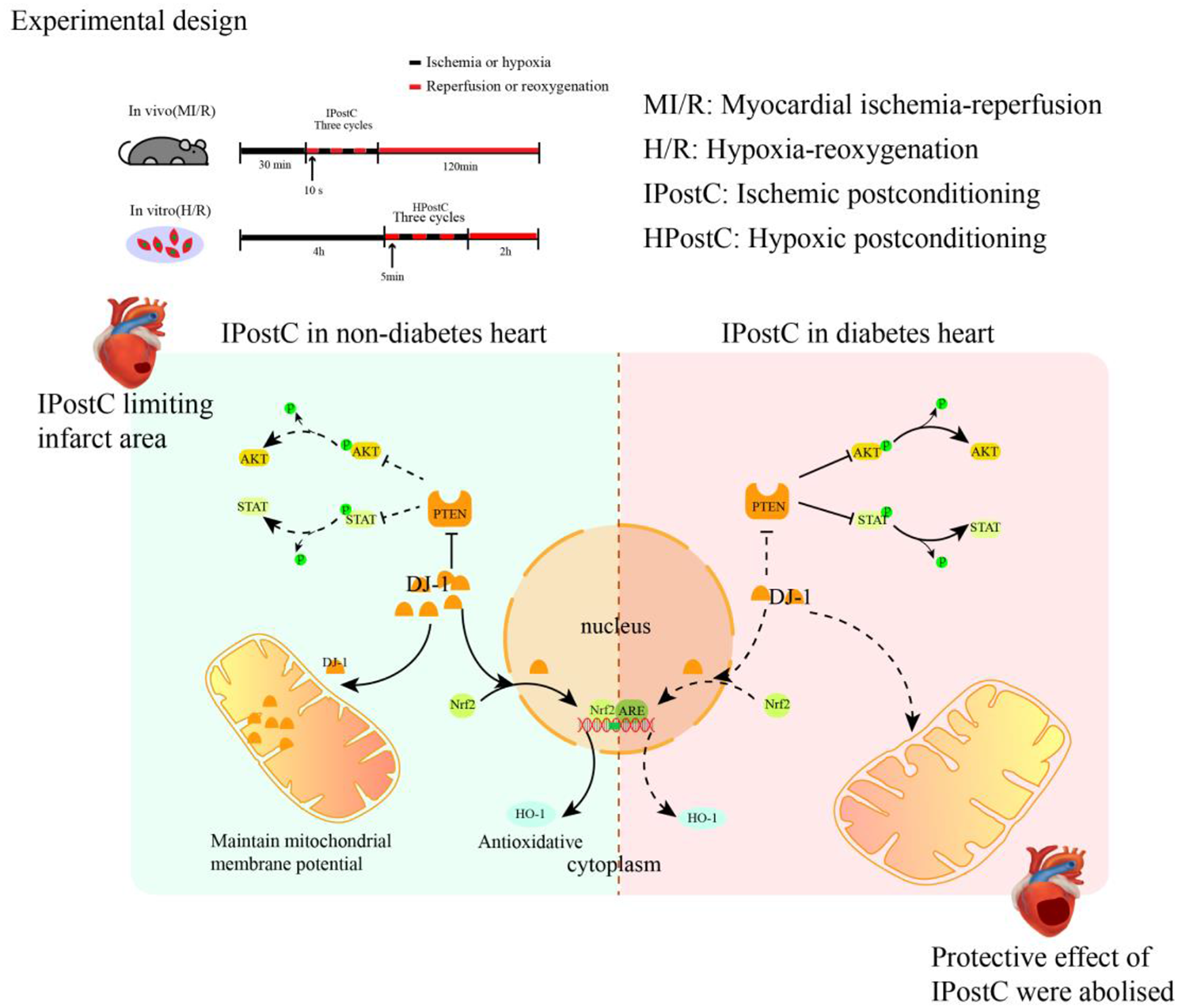
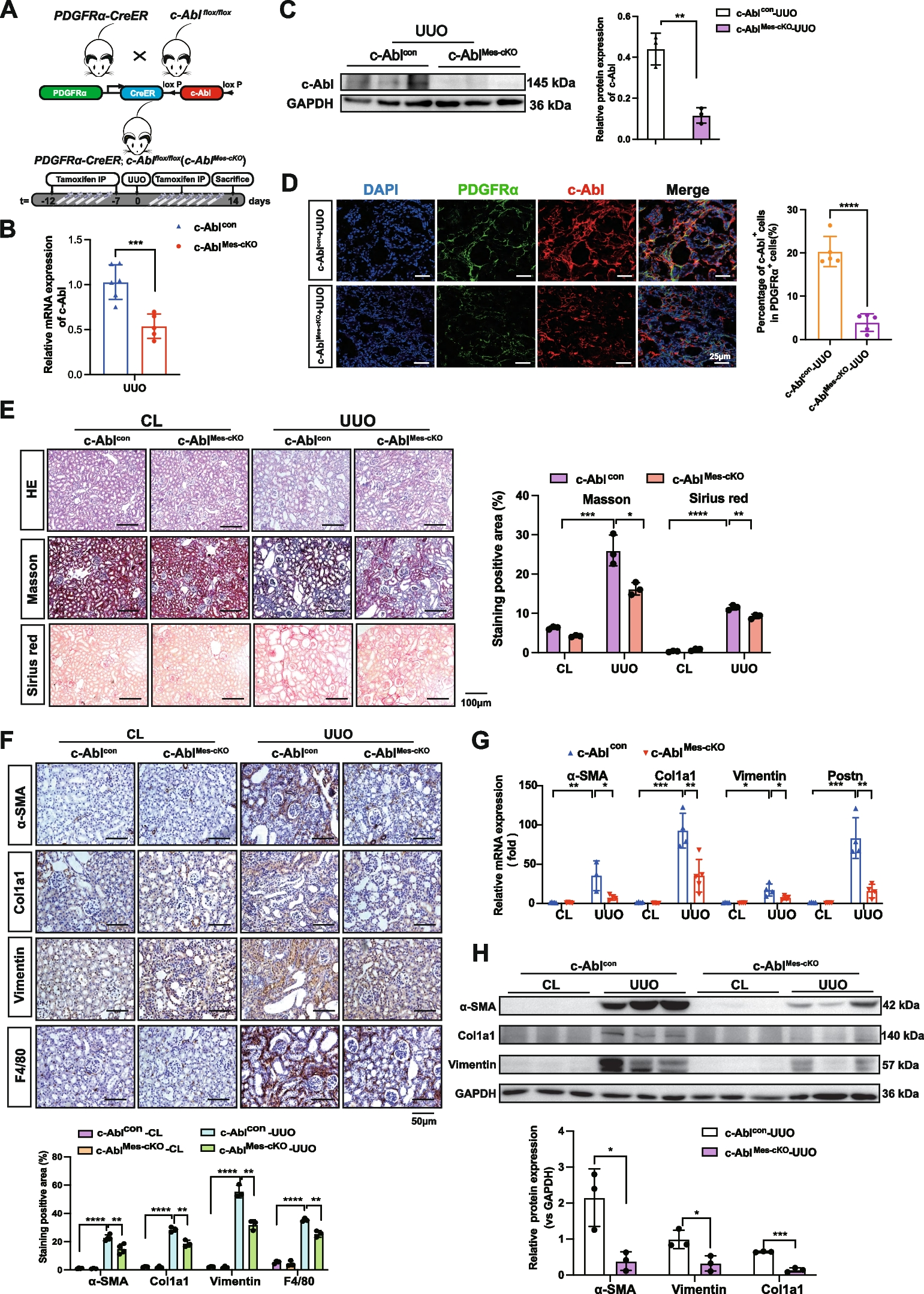
Since Gαt1 and Gαq share approximately 60% homology with Gαi1 but do not interact with AC, they have been proven as suitable partners for generating chimeras with Gαi subunits [17, 27]. A series of Gαi1 chimeras were constructed (Fig. 2A) with one or more of their putative effector recognition domains substituted by homologous regions of Gαt1 (Chi1-4) or Gαq (Chi5-6). We began by swapping the entire α4 helix to the α4/β6 loop of Gαi1 (residues 297–318) with the homologous region of Gαt1 to form Chi1 (referred to as Chi3 in [27]) (Fig. 2A). This domain was previously demonstrated to be important for AC inhibition by Gαi2 [17, 18] and Gαz [19]. Chi2 was created by an additional swapping in the switch III region (referred to as Chi7 in [27]). This chimera was found to interact with phosphodiesterase γ (PDEγ) as efficiently as Gαt1 [27], and therefore may have switched its effector preference from AC into PDEγ. Chi3 was constructed with the Gαt1 sequence in Chi1 extended up to the C-terminus (Fig. 2A) because an equivalent chimera (named as zt295) using GαzQL as the backbone resulted in a loss of the constitutive AC inhibition [19]; the AC-inhibiting surface of Gαi1 might be similarly affected in Chi3. Chi4 (also referred to as Chi4 in [27]), was designed such that both the switch III region and the C-terminal region starting from the α4 helix of Gαi1 were swapped with that of Gαt1 (Fig. 2A). Similar to Chi2, this chimera was previously shown to interact with PDEγ, which suggests that the effector specificity of the Chi4 is geared towards PDEγ [27]. Chi5 and Chi6 were equivalent to Chi1 and Chi3, except Gαq sequence was used to replace the targeted segments of Gαi1 (Fig. 2A). As Gαq has a lower overall homology to Gαi1 than Gαt1 [29], it is expected that such replacement would be more effective than Gαt1 in abolishing the activity of the GTPase-deficient mutations. In addition, Chi6 has retained the last 5 residues of the Gαi1. Retainment of the last 5 residues of Gαi would allow subsequent examination of the chimera for activation by Gi-coupled receptor [32].
Fig. 2
Construction and expression of Gαi1 chimeras. A Homologous replacements or point mutations on putative effector-interacting domains were made between Gαi1 (black) and Gαt1 (orange) (Chi1-4) or Gαq (green) (Chi5 and Chi6). Sites of replacement/mutation are indicated by their residue numbers. The locations of GTPase-deficient mutations, namely R178C and Q204L, and PTX-insensitive mutation (C351I) are highlighted with yellow dots. B Expression of the chimeras was verified by Western blotting. HEK293 cells in a 24-well plate were transfected with 0.2 μg of various chimeric constructs and the cell lysates were subjected to immunoblotting using antibodies against Gαi1 and β-actin. Expressions of the chimeras were compared with that of Gαi1
We have additionally incorporated several point mutations that have previously been found to be important for effector interactions in selected chimeras (Figs. 1B and 2A). Two residues on the α3 helix of Gαt1 (H244 and N247; equivalent to K248 and D251 in Gαi1) are critical albeit not sufficient for conferring its activity [28], but full activity can be attained in association with another residue (F283) on the αG/α4 loop [21]. Since this latter residue is also critical for the stimulatory activity of Gαs [21], it may represent an important determinant for interaction between Gαt1/PDEγ and Gαs/AC. Unlike Gαt1 and Gαs, Gαi1 possesses the more polar Y287 at the corresponding location (Fig. 1B). Hence, combinatorial replacement of K248, D251, and Y287 by cognate residues of Gαt1 (Fig. 2A) may impair the AC-inhibiting ability of the Gαi1/t1 chimeras. Another study on the effector-interacting domain of Gαq revealed the importance of three consecutive residues in the switch III region (DNE motif, homologous to EEM in residues 238–240 of Gαi1) [33]. Owing to a conserved structure across all Gα subunits, it is possible that AC interaction will be eliminated when these three residues on Chi1 are all substituted by alanine (resulting in Chi1-AAA; Fig. 2A). All chimeras were expressed at levels comparable to parental Gαi1 in transiently transfected HEK293 cells (Fig. 2B).
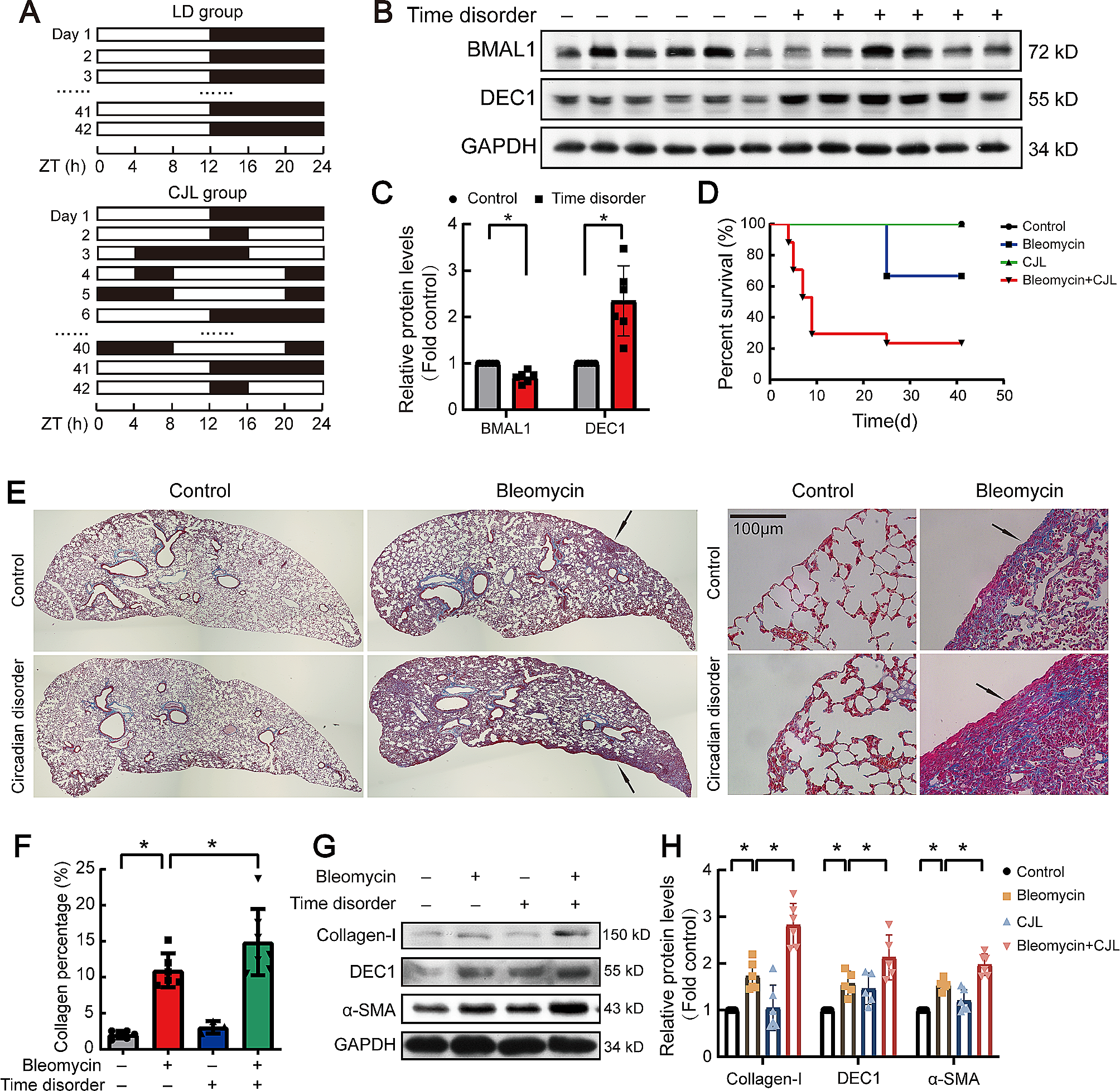
Constitutive activity of Gαi1RC is abolished by replacement of putative AC-interacting domains of Gαi1To test the effects of substitutions/mutations on the function of Gαi1, chimeras with or without either a QL or RC mutation were transfected into HEK293 cells, followed by the measurement of forskolin-induced [3H]cAMP accumulation. Three chimeric constructs, namely Chi1-KDY, Chi2-KDY and Chi6, showed constitutive stimulation/inhibition of AC activity without the incorporation of QL or RC mutations (Fig. 3A). Both Gαi1QL and Gαi1RC mutants suppressed cAMP elevation by forskolin to approximately 60% of the level observed with Gαi1 (Fig. 3B and C), consistent with previous findings indicating their constitutive activity [34,35,36]. Interestingly, as compared to the wild-type chimeras, none of the substitutions with Gαt1 affected the ability of the QL chimeras to inhibit AC (Fig. 3B). Yet, most of the RC chimeras (except Chi4RC) have lost the ability to inhibit cAMP production (Fig. 3C). It is noteworthy that purified Chi2 and Chi4 (referred to as Chi7 and Chi4 respectively in [27]) bind PDEγ as efficiently as an activated Gαt1 [27], but Chi2QL and Chi4QL/RC remained able to inhibit AC when overexpressed in cells (Fig. 3B). Our findings clearly showed functional differences between Gαi1QL and Gαi1RC (albeit both are constitutively active) in cellulo. Apparently, the activity of Gαi1RC can be more easily compromised by chimeric manipulations. A summary of their inhibitory activities towards AC is shown in Table 1.
Fig. 3
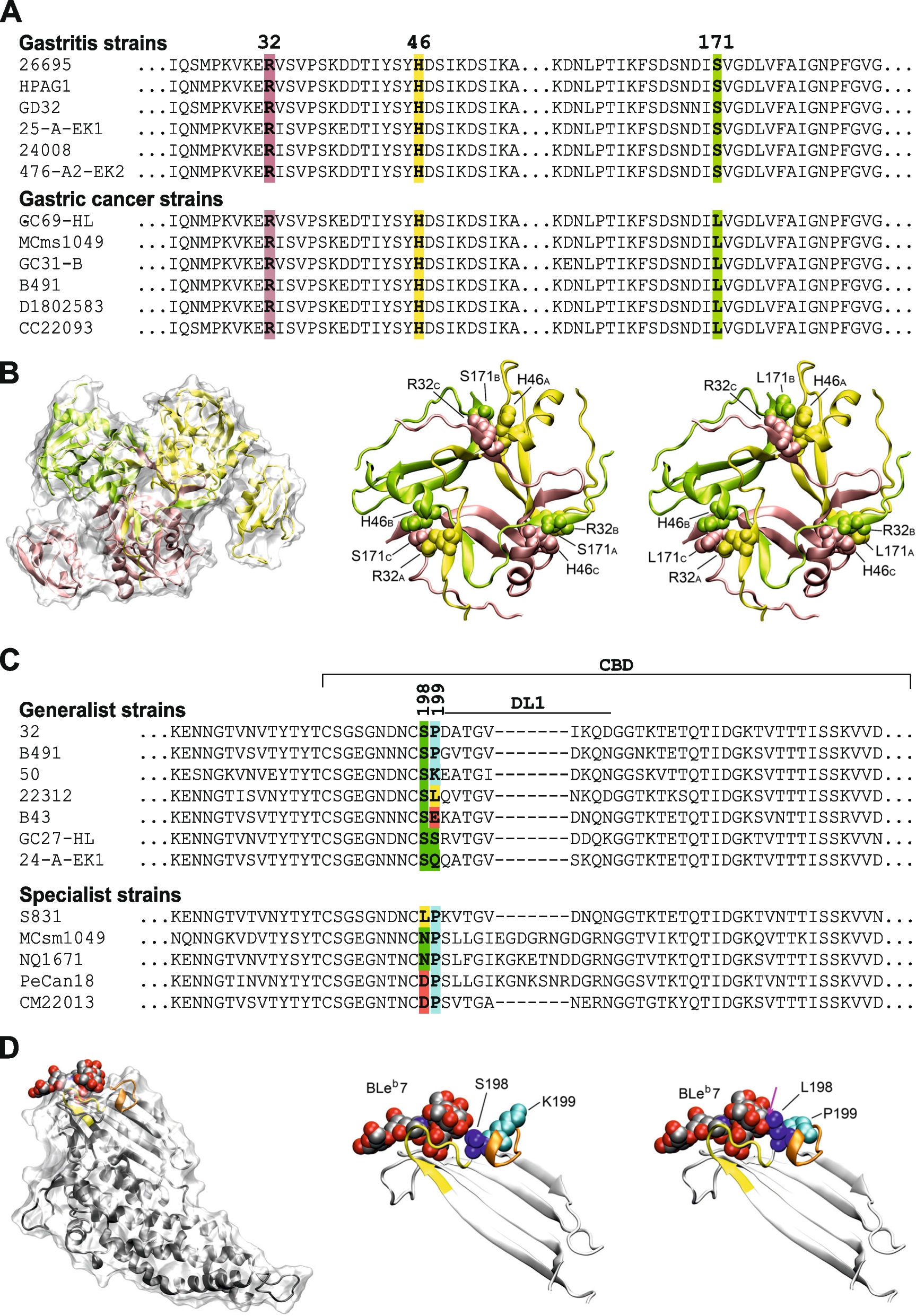
Effect of the QL/RC-bearing Gαi1 chimeras on forskolin-induced cAMP accumulation. HEK293 cells were transfected with 0.4 μg/mL of various chimeric constructs, labeled with [3H]adenine, and then assayed for [3H]cAMP accumulation in the presence of 50 μM forskolin. A Responses of the chimeras in WT version, as well as cells transfected with empty vector control (gray bar), towards forskolin were normalized against that of Gαi1. *, significantly lower than Gαi1; #, significantly higher than Gαi1. B, C The relative activities of the QL (B) or RC (C) chimeras are expressed as a percentage of cAMP accumulation of their corresponding WT. *, significantly lower than the corresponding WT, #, significantly higher than the corresponding WT. Data shown are mean ± SEM (n = 3). Bonferroni t test, p < 0.05
Table 1 Activities of QL-/RC-bearing chimeras towards forskolin responseChi6 appeared to inhibit AC constitutively (Fig. 3A). As the C-terminus of Gαq is important for effector interaction [33], we sought to test if its effector specificity has been switched to PLCβ which may then indirectly inhibit AC activity [37]. Chi6QL did not stimulate the production of inositol phosphates (IP) whereas constitutively active GαqQL significantly stimulated the PLCβ activity under the same experimental condition (Fig. S1A), suggesting that Chi6 cannot activate PLCβ.
Activity-compromised Gαi1 chimeras can suppress cAMP level upon receptor activationIn the preceding experiments, many RC-bearing chimeras lost their ability to inhibit AC while most of the chimeric QL mutants remained able to suppress the forskolin response (Fig. 3 and Table 1). The contrasting results obtained with the QL and RC mutants of the chimeras implied that there may be discernable differences in the active conformations promoted by these two mutations. We thus examined which of the two mutants have a closer resemblance to Gαi1 activated by a receptor, with the latter being more biologically relevant. We determined the chimeras’ ability to mediate receptor-induced inhibition of cAMP accumulation. To enable detection of receptor-mediated responses without interference from endogenous Gi proteins, a C351I (CI) mutation was introduced into the chimeras to provide resistance to PTX [38]. Eight chimeras that exhibited differential abilities to abolish the constitutive activities of the QL or RC mutation were selected and their corresponding CI mutants constructed; with the exception of Chi5-CI, these chimeras were expressed at levels comparable to that of the Gαi1-CI mutant (Fig. 4A). HEK293 cells co-expressing the Gi-coupled dopamine D2 receptor (D2R) and a chimera with the CI mutation were pretreated with PTX before assaying for forskolin-induced cAMP accumulation in the absence or presence of 100 nM of quinpirole (agonist for D2R). PTX treatment effectively inhibited the ability of Gαi1 to be activated by D2R (Fig. 4B), hence any detected suppression of cAMP level would be primarily due to the activity of the PTX-resistant chimeras. The positive control, Gαi1-CI, produced ~ 60% inhibition of forskolin-induced cAMP response upon activation by the receptor (Fig. 4B and C). Surprisingly, all CI chimeras significantly inhibited AC upon D2R activation (Fig. 4B), albeit weaker than that of Gαi1-CI (Fig. 4C). The extent of inhibition varied among the chimeras, with a maximum of 50% inhibition observed with Chi3-CI, while Chi1-AAA-CI and Chi5-CI only produced ~ 20% inhibition (Fig. 4C). Chi1-AAA-CI, Chi2-CI and Chi3-CI had an elevated cAMP level upon treatment with forskolin, ranging from a 30% to 50% increase (Fig. 4B).
Fig. 4
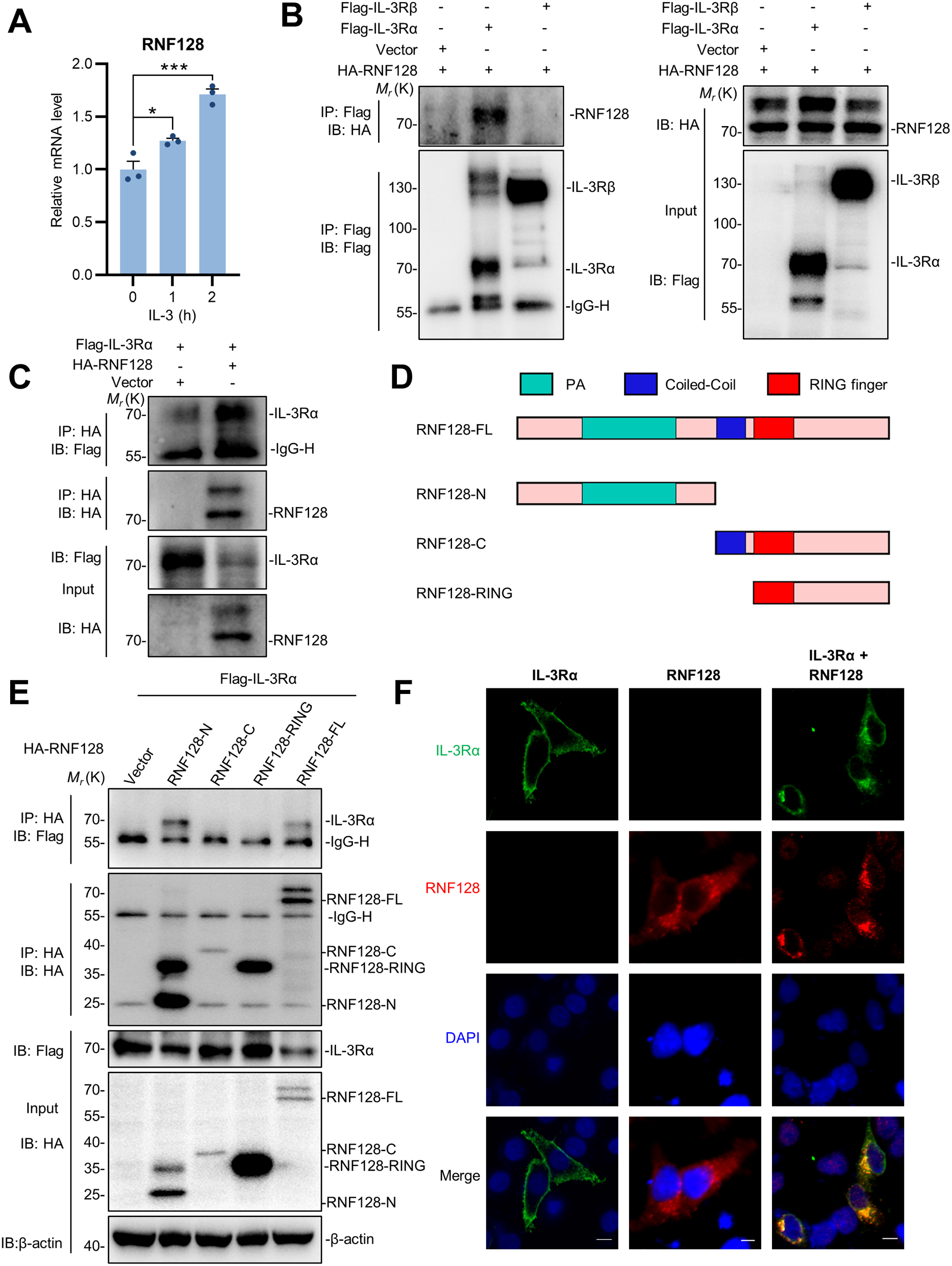
Activity of PTX-insensitive Gαi1 chimeras upon receptor activation. HEK293 cells were co-transfected with D2R and various Gαi1 constructs (0.2 μg/mL each), treated with PTX (100 ng/mL, 16 h), and then assayed for forskolin-induced [3H]cAMP accumulation in the absence or presence of 100 nM quinpirole. A Expression of the PTX-insensitive Gαi1 chimeric mutants was confirmed by immunoblotting with 20 μg of total protein. B Forskolin-stimulated cAMP levels are expressed as a percentage of the response normalized against Gαi1-CI. C Quinpirole-induced activity is expressed as a percentage of inhibition of the forskolin response. Data shown are mean ± SEM (n = 3). Bonferroni t test, p < 0.05; *, significantly lower than the control; #, significantly higher than the control; †, significant inhibition upon receptor activation. D Rationale of the subunit dissociation assay. Activated Gαi1 dissociates with Gβγ, resulting in a drop in Gαi1 intensity in immunodetection after co-immunoprecipitation with the Flag-tagged Gβ. Gαi1 activation by GTPγS, but not AlF4−, requires guanine nucleotide exchange. E–G HEK293 cells were transiently co-transfected with 0.2 μg/mL each of Flag-tagged Gβ1, HA-tagged Gγ2, and either vector (V), Gαi1 or Chi1. E Expressions of the G proteins were confirmed by immunoblotting with 20 μg of the total proteins. F 500 μg of the total proteins of the lysate were incubated with or without AlF4− (30 μM AlCl3 plus 10 mM NaF) or 100 μM of GTPγS at 37 °C for 15 min prior to immunoprecipitation by anti-Flag affinity gel. G Quantification of the co-immunoprecipitation results. Results are expressed as a percentage of Gαi1 or Chi1 pull-down by Flag-Gβ1. Graph is shown as mean ± SEM (n = 3). Student t test, p < 0.05; †, significantly different
Because GDP/GTP exchange on the Gα subunit triggered by an activated receptor is initiated from the C-terminal end of the Gα subunit to the switch regions [39], alterations in the C-terminal half of Gαi1, as in the chimeras, may affect the rate of guanine nucleotide exchange, thereby attenuating its ability to inhibit AC. To test if Chi1, a prototypical chimera, can adopt the active conformation as efficiently as Gαi1, we examined GTP-induced release of Gβγ in HEK293 cells co-expressing Flag-tagged Gβ1 and HA-tagged Gγ2 with Chi1 or Gαi1 (Fig. 4D). Lysates were treated with either aluminum fluoride (AlF4−) or GTPγS to activate the Gα subunits. AlF4− acts as a mimetic of the γ-phosphate of GTP in GDP•AlF4−-bound Gα subunits, and it can thus activate Gα subunits without requiring guanine nucleotide exchange (Fig. 4D) [40]. GTPγS is a non-hydrolyzable analog of GTP which locks the Gα subunit into an active conformation upon guanine nucleotide exchange (Fig. 4D) [41]. Activated Gαi1 should dissociate from the Gβγ dimer and thus would not co-immunoprecipitate with the Flag-tagged Gβ1 subunit (Fig. 4D). Expression of the different G protein subunits in the transfectants was confirmed by Western blots (Fig. 4E). The HA-tagged Gγ2 was efficiently co-immunoprecipitated with Flag-Gβ1, in line with Gβγ being a constitutive dimer in cells. As shown in Fig. 4F (lanes 5 and 8), both Gαi1 and Chi1 were pulled down by anti-Flag affinity beads along with the Flag-tagged Gβ1 subunit. Upon treatment with GTPγS, almost all Gαi1 dissociated from the Gβγ dimer (Fig. 4F, lane 6), but a substantial portion of Chi1 remained associated with the Gβγ dimer (Fig. 4F, lane 9); the extent of co-immunoprecipitation was quantified in Fig. 4G. In contrast, AlF4− treatment resulted in the dissociation of ~ 60% of the Gβγ-bound Gαi1 and Chi1, suggesting that Chi1 can adopt an active conformation similar to Gαi1 (Fig. 4F and G). Since the effect of GTPγS requires the release of bound GDP from the Gα subunit while the action of AlF4− is independent of such an event, these results indicate that the rate of guanine nucleotide exchange of Chi1 may be impaired, leading to apparent reductions in the AC inhibitory activity of the chimeras. This also implies that the loss of activity of RC chimeras is not due to their inability to interact with the downstream effector. Instead, the GTPase deficiency brought about by RC mutation is compromised.
Although Chi5 showed no inhibitory effect on cAMP level (Fig. 3A), Chi5-CI appeared to constitutively inhibit the forskolin-stimulated cAMP accumulation (Fig. 4B), and the forskolin response was further suppressed upon D2R-induced activation of Chi5-CI (Fig. 4C). Given that Chi5-CI contains the PLCβ-activating domain of Gαq [33], we examined if this chimera could generate IP3/Ca2+ signals via Gq. Quinpirole-induced IP formation was readily observed with Gαqz5 (positive control) [32] but not with Chi5-CI (Fig. S2A). Gαqz5 also showed a typical dose–response curve on Ca2+ mobilization upon D2R stimulation, with the maximum signal observed at 100 nM quinpirole (Fig. S2B). Yet, Chi5-CI did not stimulate Ca2+ mobilization even at 10 μM quinpirole (Fig. S2B). Therefore, Chi5-CI did not stimulate the Gq signaling pathway.
Gαi1RC-CI can respond to receptor activationThe ability of D2R to activate CI-bearing chimeras and suppress the forskolin response (Fig. 4) indicated that these chimeras still contain the necessary domains for interacting with AC. This also explains the inhibitory actions as observed with the chimeric QL mutants (Fig. 3 and Table 1). The lack of constitutive activity of the corresponding RC mutants, however, suggested that the active conformation of these Gαi1 chimeras cannot be efficiently induced and/or maintained. Hence, we asked if Gαi1QL and Gαi1RC would respond differently to receptor activation. The CI mutation was introduced into Gαi1QL and Gαi1RC and the resultant mutants, named as Gαi1QL-CI and Gαi1RC-CI, were co-expressed with D2R in HEK293 cells and then subjected to PTX treatment prior to assaying for forskolin-stimulated cAMP accumulation. In the absence of quinpirole, Gαi1QL-CI significantly suppressed the cAMP level to 50% of that obtained with the control (Gαi1-CI; Fig. 5A). This constitutive activity of Gαi1QL-CI was similar to that of Gαi1-CI-mediated AC inhibition upon D2R activation by quinpirole, indicating attainment of maximal inhibitory activity. However, cells co-transfected with D2R and Gαi1RC-CI produced an unexpected 20% increase in the forskolin response (Fig. 5A). In the presence of quinpirole, Gαi1RC-CI significantly inhibited the forskolin response by over 55% (Fig. 5A), thus suggesting that Gαi1RC-CI can interact with the receptor. This observation is important because it eliminates several possibilities that might account for the loss of AC-inhibitory ability of Gαi1RC-CI when co-expressed with D2R. Firstly, as the PTX-insensitive mutants showed similar expression levels (Fig. 5B), the lack of AC inhibition by Gαi1RC-CI was not attributed to decreased expression of this mutant. Secondly, the ability of quinpirole-treated Gαi1RC-CI-expressing cells to suppress forskolin-induced cAMP elevation to a level similar to Gαi1-CI upon receptor activation (Fig. 5A) suggested that Gαi1RC-CI can adopt an active conformation, allowing its interaction with AC. As Cys-351 is distant from the nucleotide binding pocket of Gαi1RC [42], it is unlikely that the CI mutation would directly participate in GTP hydrolysis to inactivate Gαi1RC-CI.
Fig. 5
RC mutants can be activated by receptor and suppressed by RGS. A HEK293 cells were co-transfected with D2R and various Gαi1-CI or Chi1-CI mutants and assayed similarly to Fig. 4B. The forskolin-stimulated cAMP levels of the chimeras with a CI mutation are expressed as a percentage of the response obtained with Gαi1-CI. Data shown are mean ± SEM (n = 3). Bonferroni t test, p < 0.05; *, significantly lower than the control; #, significantly higher than the control; †, significant inhibition upon receptor activation. B Expression of the PTX-insensitive mutants was confirmed by immunoblotting with 20 μg of total protein. C-E HEK293 cells were transfected with QL-bearing Gαi1 constructs and assayed similarly to Fig. 3. C Relative activities of the constitutively active chimeras are expressed as a percentage of cAMP accumulation of their corresponding WT. *, significantly lower than the corresponding WT; #, significantly higher than the corresponding WT. Data shown are mean ± SEM (n = 3). Bonferroni t test, p < 0.05. D Responses of the chimeras in WT version towards forskolin were normalized against that of Gαi1. E Expression of Gαi1 constructs were confirmed by immunoblotting with 20 μg of total protein. F–H HEK293 cells were transiently co-transfected with Flag-Gβ1, HA-Gγ2, and with or without various Gαi1 constructs and assayed by subunit dissociation assay as in Fig. 4D. F Expressions of the G proteins were confirmed by immunoblotting with 20 μg of the total proteins. G 500 μg of the total proteins of the lysate were incubated with or without 100 μM of GTPγS at 37 °C for 15 min prior to immunoprecipitation by anti-Flag affinity gel. H Quantification of the co-immunoprecipitation results. Results are expressed as a percentage of the corresponding Gαi1 or Chi1 constructs pull-down by Flag-Gβ1. Graph shown as mean ± SEM (n = 3). Student t test, p < 0.05; n.d., not detectable; ns, non-significant; #, significantly higher than the control
Next, we examined if the loss of activity of RC chimeras is due to their failure to maintain, or alternatively, induce the active conformation of the Gα subunit. To test this, we introduced the CI mutation to Chi1QL and Chi1RC, the prototypical chimeric constructs. Chi1-CI showed approximately 25% suppression of cAMP level upon receptor activation (Fig. 5A). Chi1QL-CI was constitutively active without quinpirole treatment, with the forskolin response reduced to a level similar to an activated Chi1-CI (Fig. 5A). Receptor activation enhanced the inhibition on cAMP level by Chi1QL-CI, suggesting Chi1QL-CI is not fully active (Fig. 5A). Like Gαi1RC-CI (Fig. 5A), Chi1RC-CI did not inhibit the forskolin-induced cAMP accumulation and showed prominent AC inhibition only upon quinpirole treatment, indicating that the active conformation of Chi1RC-CI is inducible (Fig. 5A). Thus, the loss of AC inhibition by RC chimeras may be attributed to the lack of maintenance of their active conformation.
Chi1RC is RGS-sensitive in celluloSince Gαi1RC-CI and Chi1RC-CI could be activated by D2R (Fig. 5A), it implies that they may adopt an inactive conformation in the absence of receptor activation despite harboring the RC mutation. Because an active GTP•Gαi1 has a low affinity for the receptor [43], it further suggests that a substantial portion of the Gαi1RC-CI is GDP-bound. Given that the RC mutation impairs the intrinsic GTPase activity [44], the GDP-bound state (as opposed to a GTP-locked state) can be obtained by two means: the prevention of GDP/GTP exchange by guanine nucleotide dissociation inhibitors, and the extrinsic promotion of GTP hydrolysis by GTPase-activating proteins (GAPs). An early reconstitution study showed that RGS4 could promote the GTP hydrolysis of Gαi1RC, but not for Gαi1QL [24], although in cellulo evidence remains lacking. Thus, the lack of constitutive activity of RC chimeras may be attributed to their interaction with RGS proteins which aids in maintaining the GDP-bound state of the Gα subunits. To test this hypothesis, we incorporated an RGS-insensitive G183S mutation [45] into QL/RC-bearing Gαi1 and Chi1, and then examined their AC inhibitory activities. As shown in Fig. 5C, both QL and RC versions of Gαi1-G183S constitutively suppressed cAMP accumulation to an extent similar to Gαi1QL and Gαi1RC, respectively. It is also worth noting that G183S mutation alone did not produce any effect on AC inhibition (Fig. 5D). These observations suggested that RGS proteins did not hinder the interactions between AC and the two constitutively active mutants. Both Chi1QL and Chi1QL-G183S produced significant AC inhibition (Fig. 5C). Strikingly, G183S mutation enabled Chi1RC to suppress cAMP production (Fig. 5C). Although mutants bearing the G183S mutation showed a lower expression (Fig. 5E), the level was nevertheless sufficient to generate a significant cAMP suppression (Fig. 5C). Collectively, the lack of AC inhibition by Chi1RC, and possibly other RC chimeras, might be attributed to ‘hyper’-interactions of the Gα subunits with RGS proteins. This also provides the first in cellulo evidence that RC mutation is RGS-sensitive.
As Chi1RC-G183S inhibits AC to the same extent as Gαi1RC (Fig. 5C), one would expect activated Chi1-G183S to dissociate from the Gβγ dimer like a Gαi1. However, it is also possible that RGS proteins may displace the Gβγ dimer from an active Gαi1. Co-crystal structures of RGS-Gαi1 reveal that RGS proteins bind orthogonally to the switch regions of Gαi1 [25, 46]. In fact, RGS4 inhibited Gαq-mediated activation of PLCβ1 by direct blockade of the binding interface [
留言 (0)