
記住我






Fibrosis induced by proliferation of extracellular matrix (ECM) components, such as collagen and fibronectin, in addition to regeneration, is essential for injury repair. Physiologically, the proliferated connective tissue effectively promotes the repair of injury, whereas excess components of the ECM are rapidly removed in a short period. However, persistent injury and chronic inflammation cause excessive accumulation of ECM components, resulting in the overproduction of scar tissue and destruction of normal tissue structure, organ dysfunction, and ultimately organ failure; this is termed pathological fibrosis. Infiltrating inflammatory cells, activated endothelial cells, and platelets at the site of injury cause inflammation, which triggers fibroblast recruitment and proliferation through the secretion of pro-fibrotic cytokines (i.e., interleukin [IL]-1 and tumor necrosis factor alpha [TNF-α]) and various growth factors, such as transforming growth factor beta (TGF-β), platelet-derived growth factor (PDGF), epidermal growth factor (EGF), and fibroblast growth factor (FGF). TNF-α, TGF-β, and PDGF also promote fibroblast activation and differentiation into myofibroblasts, resulting in the synthesis of more collagen for extracellular accumulation. More important for the excessive collagen accumulation is the inhibition of its degradation in this environment, which—together with overproduction—leads to pathological fibrogenesis [Figure 1A].[1] However, there is a lack of effective approaches to prevent or reverse fibrosis.[1]
 Figure 1:
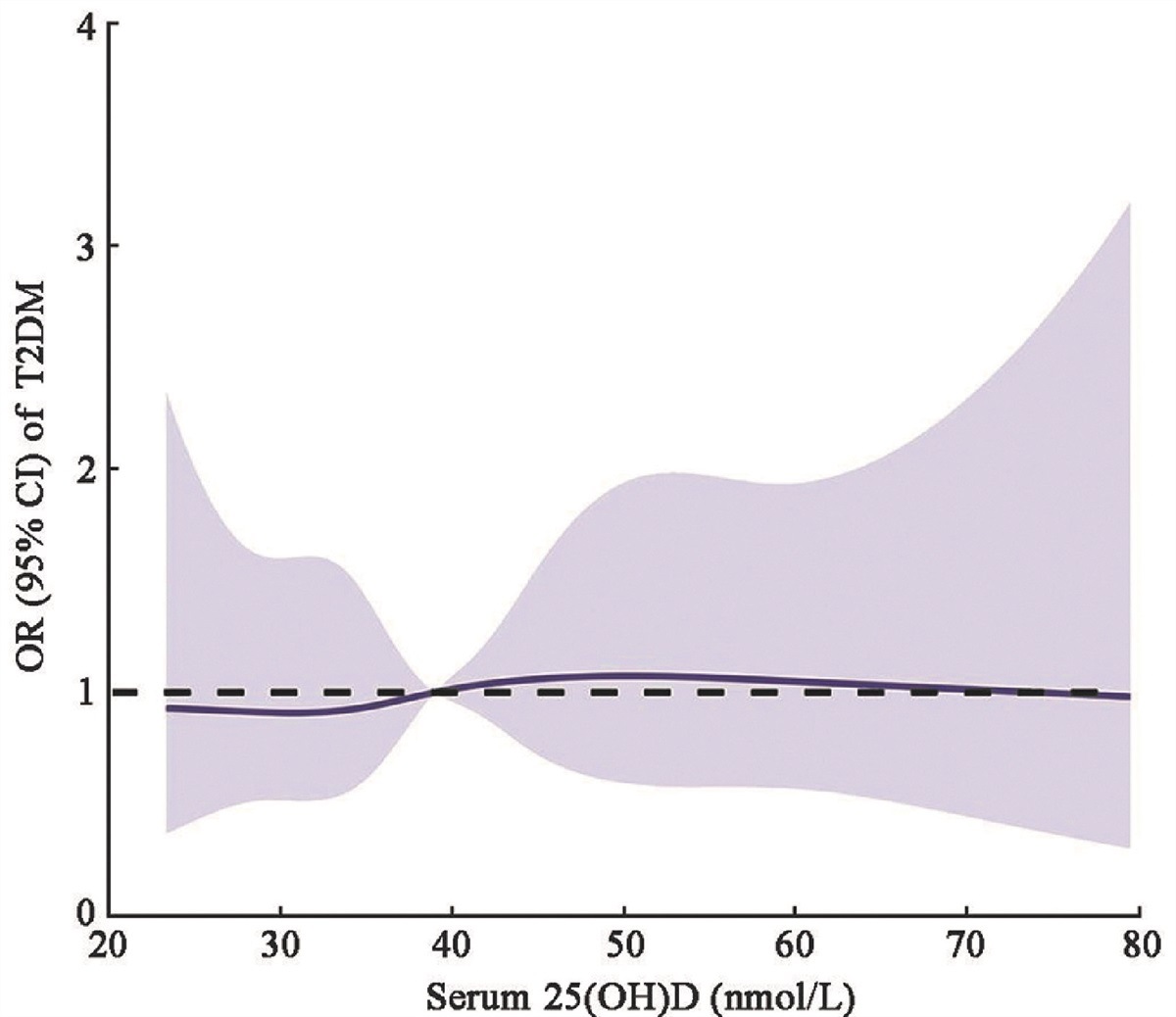
Figure 1: Mechanisms of fibrosis (A) and ferroptosis (B), and the role of ferroptosis in fibrosis. Ferroptosis facilitates fibrosis by five primary mechanisms involving "exacerbating tissue injury," "recruiting inflammatory cells and evoking necroinflammation," "releasing pro-fibrotic factors," "facilitating the activation of myofibroblasts," and "promoting EMT." ACSL4: Acyl-coenzyme A synthetase long-chain family member 4; ALOX: Arachidonate lipoxygenase; CD98: Cluster of differentiation 98; CoA: Coenzyme A; ECM: Extracellular matrix; EMT: Epithelial–mesenchymal transition; EGF: Epidermal growth factor; FGF: Fibroblast growth factor; FPN: Ferroportin; FTH1: Ferritin heavy chain 1; FTL: Ferritin light chain; GLS: Glutaminase; GPX4: Glutathione peroxidase 4; GSH: Glutathione; GSR: Glutathione reductase; GSSH: Oxidized glutathione; IL-1: Interleukin-1; LPCAT3: Lysophosphatidylcholine acyltransferase 3; LPO: Lipid peroxidation; NADPH/NADP+: Nicotinamide adenine dinucleotide phosphate; NCOA4: Nuclear receptor coactivator 4; NOX: NADPH oxidase; PDGF: Platelet-derived growth factor; PL-PUFA: Polyunsaturated phospholipid; PUFA: Polyunsaturated fatty acid; PUFA-OH: Phospholipid hydroxide; PUFA-OOH: Phospholipid hydroperoxide; ROS: Reactive oxygen species; SLC1A5: Solute carrier family 1 member 5; SLC3A2: Solute carrier family 3 member 2; SLC40A1: Solute carrier family 40 member 1; SLC7A5: Solute carrier family 7 member 5; SLC7A11: Solute carrier family 7 member 11; STEAP3: Six-transmembrane epithelial antigen of prostate 3; TF: Transferrin; TfR1: Transferrin receptor 1; TGF-β: Transforming growth factor beta; TNF-α: Tumor necrosis factor alpha. Parts of the figure were drawn by using or modifying pictures from Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 Unported License.
A decade has passed since ferroptosis, a process of iron-driven lipid peroxidation (LPO)-mediated programmed cell death, was proposed.[2] Over time, scientists have increased their understanding of ferroptosis. Acyl-coenzyme A synthetase long-chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3) activate and incorporate polyunsaturated fatty acids (PUFAs), such as arachidonic acid, into membrane-localized lipids (i.e., phospholipids). In the absence or inhibition of the cystine/glutamate antiporter system (system xc-) and/or glutathione peroxidase 4 (GPX4), these PUFA-containing membrane lipids undergo LPO driven by labile iron and iron-dependent enzymes (i.e., arachidonate lipoxygenases [ALOXs]).[3]
Ferroptosis has been linked to various disease processes, such as cancers, ischemia-reperfusion injury (IRI), neurodegeneration, autoimmune diseases, and infections.[3] Furthermore, recent studies have reported that ferroptosis is associated with fibrogenesis in multiple diseases. The links between different types of cell death and fibrosis have always been a focus of researchers. Parenchymal cell death through apoptosis, pyroptosis, and necroptosis[4] has been reported to function as an initiator of fibrosis. It elicits an inflammatory response in which resident immune cells or hemopoietic cells enter the affected region, secrete pro-inflammatory cytokines and chemokines, transform from a resting to an activated phenotype, and synthesize ECM components. The number of studies on ferroptosis in fibrotic diseases is also increasing. Therefore, this article aims to provide a review of the evidence for the involvement of ferroptosis in fibrosis and the potential for ferroptosis-based therapies for the treatment of fibrosis.
Mechanisms of Ferroptosis and Iron MetabolismFirst described in 2012, ferroptosis was discovered through the identification of RAS-selective lethal molecules (RSLs) that unexpectedly induced a unique form of cell death distinguished from apoptosis, necrosis, and autophagy.[2] It is characterized by shrunken mitochondria with increased membrane density (morphologically), depletion of glutathione (GSH) and inactivation of GPX4 (biochemically), and alteration of iron, amino acid, and lipid metabolism-related genes (genetically).[2]
The late event of ferroptosis features the peroxidation of membrane-bound, rather than free PUFAs with a compromised antioxidant mechanism in cells.[3] Research has demonstrated the central role of two regulatory factors, namely ACSL4 and LPCAT3, in regulating the process of lipid activation and accumulation as part of the peroxidation process. Inhibition of these enzymes has been found to confer resistance to ferroptosis.[5] Other ACSL enzymes are also associated with lipid generation and incorporation into cell membranes. For example, ACSL1 promotes α-eleostearic acid incorporation into neutral lipids.[3] Furthermore, ACSL3 is required for the activation of exogenous monounsaturated fatty acids and their incorporation into phospholipids, where they displace PUFAs and exert anti-ferroptotic effects.[3]
The critical antioxidant mechanism that confers resistance to ferroptosis is the system xc-/GSH/GPX4 pathway, which plays a crucial role in the maintenance of cellular oxidative homeostasis. System xc- is composed of solute carrier family 7 member 11 (SLC7A11, also known as xCT) and SLC3A2 subunits, which export glutamate and import cystine. The cystine is then reduced to cysteine and is used to generate GSH, which functions as an electron donor in the activity of the selenoprotein GPX4, a critical enzyme in the defense against oxidative species production. Studies suggest that the system xc-/GSH/GPX4 pathway is of paramount importance in the resistance to ferroptosis. Initial research reported the induction of ferroptosis upon inhibition of system xc--mediated cystine uptake by erastin, which binds to SLC7A5 or system L (the SLC7A5/SLC3A2 complex) and disrupts cystine uptake.[2] Subsequent research deepened our understanding of this mechanism and revealed that the depletion of GSH by erastin is a prerequisite for GPX4 inactivation.[6] Another potent ferroptosis-inducing agent, RSL3, has been shown to directly bind to and inactivate GPX4, and silencing of GPX4 using RNA interference (RNAi) techniques increases susceptibility to RSL3-induced ferroptosis; in contrast, the overexpression of GPX4 confers resistance to this phenomenon.[6] Free radical scavenging mechanisms that are independent of GPX4 have been shown to suppress ferroptosis by protecting against lipid reactive oxygen species (ROS) buildup. The main lipophilic radical-trapping antioxidants (RTAs) include vitamin E-OH, coenzyme Q10-H2 (CoQ10-H2 or ubiquinol), and tetrahydrobiopterin (BH4). Vitamin E-OH and CoQ10-H2 block the activation of LPO and accumulation of PUFAs containing phospholipid hydroperoxides (PL-PUFA-OOHs) through redox reactions.[3] BH4, in addition to its role as an RTA, has dual roles in generating reduced CoQ10 and rewiring the lipid membrane milieu to disfavor LPO.[3] These reactions can be enhanced by enzymatic recycling of RTAs. Ferroptosis suppressor protein 1 (FSP1, formerly known as apoptosis inducing factor mitochondria associated 2 [AIFM2]) regenerates ubiquinol from ubiquinone using reduced nicotinamide adenine dinucleotide phosphate nadph (NADPH).[3] Regulation of FSP1 involves the mouse double minute 2/10 (MDM2/MDMX) complex-peroxisome proliferator activated receptor alpha (PPARα) pathway, in which inhibition of the MDM2/MDMX complex increases PPARα level, thereby upregulating FSP1production.[7] In mitochondria, dihydroorotate dehydrogenase (DHODH) acts in parallel with FSP1 by replenishing ubiquinol.[3] Guanosine triphosphate (GTP) cyclohydrolase (GCH1) initiates the synthesis of BH2 and BH4, whereas dihydrofolate reductase (DHFR) regenerates BH4.[8] Overall, the FSP1/DHODH/CoQ10 pathway and GCH1/DHFR/BH4 pathway protect against ferroptosis by modulating LPO.
Iron is a key component of ferroptosis, as the name implies. Studies have revealed that although the intracellular lipid hydroperoxides (LOOHs) resulting from lipoxygenases (LOXs) may activate ferroptosis, it is the labile iron-facilitated LPO, an autoxidation of LOOH decomposition species (alkoxyl and/or hydroxyl radicals) generated by the Fenton reaction, that executes the ferroptosis process.[9] Dietary iron (heme and non-heme iron) taken up by duodenal enterocytes and heme iron recycled by reticuloendothelial system macrophages from aged or damaged red blood cells are the main iron sources of the organism. In the circulation, iron in the ferric (Fe3+) form binds to transferrin (TF) to form transferrin-bound iron (TBI), which is transported to organs that require iron to function. TBI binds to the transferrin receptor (TfR) on the cell membrane and is transported into the cell via endocytosis. However, if systemic iron is overloaded or TF is saturated, iron will bind to other non-transferrin proteins to form non-transferrin-bound iron (NBTI), which binds to the divalent metal transporter 1 (DMT1) to enter the cell. Intracellular Fe3+ is then reduced to the ferrous (Fe2+) by the six-transmembrane epithelial antigen of prostate 3 (STEAP3), which is further released into the cytosol. In fact, there is no free iron inside the cell; excess iron ions are either exported into the circulation through basal membrane protein ferroportin (FPN) or stored in L-, H-ferritin in a non-toxic form, making cells resistant to ferroptosis. However, a small fraction (no more than 5%) of loosely bound metabolically active ferrous iron, called labile iron, is present in the cytoplasm. Hider and Kong[10] demonstrated that GSH-Fe2+ is the predominant component of labile iron.
Evidence indicates that alterations in iron regulators contribute to ferroptosis. Nuclear receptor coactivator 4 (NCOA4) mediates the degradation of ferritin, called ferritinophagy, resulting in an increase in labile iron, which contributes to erastin-induced ferroptosis.[11] Similarly, administering miR-124toupregulate the level of iron exporter FPN rescues cell death.[12] As an important iron transporter, TF deficiency leads to ferroptosis via iron overload.[13] Interestingly, Brown et al[14] discovered that pro-ferroptotic stimuli induce the production of prominin 2, a protein implicated in the regulation of lipid dynamics, promoting the formation of ferritin-containing multi-vesicular bodies and exosomes that export iron out of cells, thereby contributing to ferroptosis resistance.
Evidence for the Involvement of Ferroptosis in Fibrosis Iron overloadThe contribution of iron dysregulation and iron deposition-mediated ferroptosis to fibrosis has been extensively documented in various organs, including the lung,[15] heart,[16] and liver.[13] It has been established that iron chelators can effectively mitigate ferroptosis-induced fibrosis.[17] A study utilizing Tf-knockout mice to simulate the TF-deficient environment observed in patients with liver cirrhosis demonstrated that iron overload-induced ferroptosis plays a role in the progression of liver fibrosis, and that the administration of a ferroptosis inhibitor or the targeting of Slc39a14 (ZRT/IRT-like protein 14 [Zip14]), a hepatic transporter for NBTI, can ameliorate fibrogenesis.[13] Moreover, alterations in iron regulation have been observed in patients with fibrosis. These changes include elevated levels of TfR1 and DMT1 on alveolar epithelial type II cells in patients with bleomycin-induced pulmonary fibrosis.[15]
Iron overload participates in the formation of fibrosis by activating pro-fibrotic pathways and increasing recruitment and activation of fibrosis-associated cells, such as macrophages. In hepatocytes, iron overload promotes fibrosis by upregulating heme oxygenase-1 (HO-1) levels, which significantly contribute to liver fibrosis. Increasing HO-1 ubiquitination and upregulating the level of nuclear factor erythroid 2-related factor 2 (Nrf2) by fibroblast growth factor 21 (FGF21), a novel oxidative stress regulator in humans, can ameliorate iron overload-induced ferroptosis, thereby protecting the liver from ferroptosis-associated fibrosis.[18] Another well-established fibrosis-mediating pathway implicated in various tissues is the TGF-β/Smad signaling pathway, which induces the activation of myofibroblasts, production of excessive ECM, and inhibition of ECM degradation. Excess iron deposition in renal tubular epithelial cells (RTECs) has been demonstrated to increase TGF-β release.[19] The application of iron chelators, such as deferoxamine (DFO), alleviates mitochondrial DNA (mtDNA) leakage and TGF-β-induced epithelial–mesenchymal transition (EMT) in the lung,[20] suggesting that iron metabolism contributes to EMT by impairing mitochondria, as iron overload-induced mitochondrial dysfunction is related to the release of mtDNA.[21]
Aberrant iron distribution may drive lipogenesis and fibrosis, the relationship between lipid metabolic dysregulation and fibrosis is also worth noting. Lipid metabolic dysregulation is observed in fibrosis and may contribute to the progression of fibrosis. Out of the context of ferroptosis, mounting evidence by using transcriptomic profiling and single-cell sequencing reveals alterations in lipid metabolism-related proteins in fibrosis. For example, adenosine triphosphate (ATP) binding cassette subfamily A member 3 (ABCA3), a transporter involved in lipid export is reduced in alveolar type II (ATII) cells during idiopathic pulmonary fibrosis (IPF)[22]; and accumulation of lipid droplets accompanied by transient increased production of lipid-droplet surface protein perilipin 2 (PLIN2) is observed in proximal RTECs (PTECs) in early stage of IRI-induced kidney fibrosis[23]. Specific mechanisms in lipid dysregulation-related fibrosis are mainly discussed around endoplasmic reticulum stress, immune response, and inflammation. For example, in non-alcoholic fatty liver disease (NAFLD), inflammatory mechanisms mediating LPO and fibrosis have been uncovered, including macrophage heterogeneity, auto-aggressive T cell, etc.[25] Association between iron overload or ferroptosis and lipid metabolic dysregulation might exist but not definite. Homeostatic iron regulator (HFE) is an iron regulatory protein (IRP) which interacts with TfR. HFE-knockout mice developed severe fatty liver disease phenotype similar to human non-alcoholic steatohepatitis (NASH) with early fibrosis via dysregulating lipid metabolism which favors lipogenesis, supported by upregulated messenger RNA (mRNA) expression of lipogenic genes.[26] However, this study lacks evidence of significant iron and LPO in Hfe-/- models, indicating that IRP can cause lipid accumulation but LPO-related fibrosis is not independent of iron overload. In addition, multiple logistic regression analysis of histological factors associated with fibrosis, comparing models with and without LPO, showed the strength of the association of iron with fibrosis weakened when counting in malondialdehyde staining, suggesting LPO at least accounted for part of the effect.[27]
On the other hand, using iron chelators or ferroptosis inhibitor (Fer-1) can deplete labile iron and LPO, thus ameliorating kidney fibrosis, indicating a possible interplay between ferroptosis and LPO in the context of fibrosis. There is also evidence of aberrant iron distribution may indirectly lead to liver lipogenesis and fibrosis progression through parenchymal–mesenchymal crosstalk. For example, the steatotic phenotype in NAFLD or NASH-afflicted liver is characterized by iron deficiency in hepatocytes and iron overload in hepatic stellate cells (HSCs) caused by iron-containing extracellular vesicles. Iron deficiency induced lipogenesis and insulin resistance via hypoxia-inducible factor-2 alpha/activating transcription factor 4 (HIF-2α/ATF4) signaling pathway. Iron accumulation in HSC exacerbates fibrogenesis by enhancing ROS production.[28] However, this study did not provide direct evidence for iron overload-induced irregular lipid distribution in promoting fibrosis.
Ferroptosis-mediated necroinflammationTissue damage induced by various stimuli, including IRI, toxins, and infections, can cause the release of damage-associated molecular patterns (DAMPs) to induce interstitial inflammation and immune response, which activates and recruits resident/infiltrating immune cells. This form of necrosis-induced inflammation is termed "necroinflammation." Ferroptosis has been reported to generate DAMPs,[29] making it a possible source of necroinflammation. In general, the inflammation that arises from tissue damage plays a role in tissue repair and the progression of fibrosis.
In studies investigating a NASH-hepatic fibrosis model, ferroptosis was shown to be the initial cell death that triggers inflammation. In mice undergoing treatment with a choline-deficient and ethionine-supplemented diet, knocking out Mlkl had no effect on the reduction of immune cell infiltration and inflammation, whereas administration of a ferroptosis inhibitor led to a considerable decrease in immune cell infiltration and levels of inflammatory factors compared with the control group.[30] These findings align with previous studies reporting that ferroptosis, not necroptosis, initiates inflammation. Inhibiting ferroptosis in acute kidney injury models improved renal function, reduced histological injury, and decreased oxidative stress.[31] Upon injury, PTECs undergo a transient pro-inflammatory state. If the initial injury is mild, the PTECs return to their original state through re-differentiation; if the injury is severe, prolonged, or recurrent, the inflammatory PTECs will de-differentiate into damage-associated PTECs (DA-PTECs), thereby contributing to persistent inflammation and a fibrotic microenvironment.[32] The molecular pathways that govern this transition may be associated with ferroptosis. Research found that DA-PTECs were susceptible to ferroptosis after severe ischemic injury and that Gpx4 deletion resulted in the accumulation of pro-inflammatory DA-PTECs and hindered renal repair after mild ischemic injury.[32] Furthermore, DA-PTECs were found to interact with resident and infiltrating myeloid cells through the enrichment of Icam1-, Pdgfb-, and Apoe-mRNA expression.[32] Sub-clustering of the infiltrating cells revealed the presence of distinct cell types following varying degrees of IRI, including cluster of differentiation (CD) 4+ T cells and natural killer cells in the control group, CD8+ T cells, Tregs, γδT cells, and dendritic cells (DCs) in the adaptively repaired kidneys (short IRI) and Ly6Chigh monocytes and granulocytes in the maladapted kidneys (long IRI).[33]
Immune cells, especially macrophages, play a pivotal role in inflammation and fibrogenesis. Macrophages have three main functions: phagocytosis, cytokine secretion, and ROS production. While research on ferroptosis–macrophage interactions is adequate, the involvement of macrophages in fibrosis warrants further investigation.
Ferroptosis in macrophages promotes polarization to the pro-inflammatory phenotype, M1. Iron loading in bone marrow-derived macrophages augments M1 polarization, possibly by reducing IL-4-mediated phosphorylation of signal transducer and activator of transcription 6 (STAT6), a transcriptional regulator of M2 activation, accompanied by an observed increase in the fibrogenic marker alpha-smooth muscle actin (α-SMA).[34] Another study reported that iron overload in macrophages promoted M1 polarization through enhanced ROS production and p53 acetylation.[35] The study initially aimed to investigate cancer immunotherapy, as M1 polarization represents an anti-tumor phenotype. Inhibiting ferroptosis in macrophages mitigated fibrosis by reducing the secretion of pro-fibrotic cytokines that promote fibroblast recruitment and activation. Specifically, treatment with Fer-1 decreased levels of IL-1β, TNF-α, and TGF-β in silicon dioxide (SiO2)-induced ferroptotic macrophages, thereby alleviating pulmonary fibrosis.[36] In addition, a study reported that FPN deficiency-induced ferroptotic hepatocytes drove macrophage proliferation and M2 polarization by increasing IL-10 and TGF-β release concomitantly with aggravated liver fibrosis.[37]
Ferroptotic cells can recruit macrophages by releasing cytokines. Monocyte chemoattractant protein-1 (MCP-1/CCL2) is a key chemokine signal involved in fibrosis across multiple organs. It is released by parenchymal cells and can activate tissue macrophages, which promote the activation of fibroblasts. In a trans-well assay, co-culturing RAW264.7 macrophages with RSL3-treated RTECs revealed elevated levels of CCL2 and enhanced macrophage migration. Transfecting with MCP-1 small interfering RNA or treatment with Fer-1, a system xc- inhibitor, reduced macrophage chemotaxis and attenuated kidney fibrosis in IRI or unilateral ureteral obstruction (UUO) models.[38] Moreover, ferroptotic cells activate macrophages by releasing DAMPs and provoke ferroptosis-evoked necroinflammation through macrophages. An in vitro study showed that ferroptotic cells elicit the autophagy-dependent release of high mobility group box 1 (HMGB1), a DAMP signal, which further triggers the release of the proinflammatory cytokine TNF from macrophages. Administration of anti-HMGB1 neutralizing antibody or knockout AGER (HMGB1 receptor) in macrophages reduced TNF production.[29] DCs respond to DAMP signals, and a study by Giuliani et al[39] found that the ferroptosis pathway plays a mediating role in inflammasome activation in human kidney DCs, leading to chronic kidney disease (CKD) progression. The study used patient-derived PTECs subjected to hypoxia to simulate the CKD microenvironment, with ferroptotic PTECs releasing putative DAMP signals to induce adjacent CD1c+ DC-derived IL-1β and IL-18 secretion through the activation of nod-like receptor (NLR) family pyrin domain containing 3 (NLRP3) inflammasomes.[39]
From another perspective, macrophages may promote ferroptosis in parenchymal cells. A review introduced the role of macrophages in facilitating ferroptosis through several mechanisms: (1) iron accumulation by phagocytosis leading to imbalanced iron metabolism in the environment; (2) IL-6-, TNF-α-, and IL-1β-mediated regulation of IRPs; and (3) production of ROS and induction of ROS-dependent ferroptosis.[40] There is evidence for this interplay in a fibrosis setting. Protein arginine methyltransferase 7 (PRMT7) is an epigenetic factor that can mono-methylate histones on arginine residues. Samples from patients with chronic obstructive pulmonary disease showed elevated PRMT7 levels. The mouse bleomycin-induced fibrosis model exhibited reduced expression of PRMT7 on macrophages, which is associated with decreased recruitment of monocytes to the injury site and less severe symptoms. Persistent monocyte-derived macrophages infiltration leads to ALOX5 overexpression, upregulation of ACSL4 level, and sensitization to ferroptosis in lung epithelial cells.[41]
Ferroptosis-related pro-fibrotic factors and parenchymal–mesenchymal interactionsThe causes of fibrosis encompass a multifarious interplay of pathological mechanisms, one of which is inflammation. However, fibrosis can also result from cell damage independently of an inflammatory response, through EMT and fibroblast–myofibroblast transition (FMT).
Interactions between ferroptosis and EMT have been extensively investigated in the field of tumor progression and cancer therapy.[42] Cells (especially cancer cells) undergoing EMT are vulnerable to ferroptosis, and epigenetic modulation of EMT contributes to enhancing ferroptosis in head and neck cancer cells.[42] However, ferroptosis also promotes EMT. The hallmarks of EMT include the downregulation of E-cadherin and upregulation of N-cadherin and α-SMA; E-cadherin marks epithelial cells, while α-SMA signifies fibroblast differentiation. In alveolar epithelial cells, application of the ferroptosis inducer erastin led to reduced E-cadherin production, indicating a tendency for ferroptosis to be involved in EMT.[43] Similarly, the inhibition of prostaglandin-endoperoxide synthase 2 (PTGS2), a driver of ferroptosis, in tumor epithelial cells led to suppression of N-cadherin and increased E-cadherin, indicating that inhibition of ferroptosis might limit EMT.[44] However, one limitation of these studies is that the experiments were mainly performed in vitro, which does not consider the in vivo microenvironment. To address this, Yao et al[45] used a single-cell RNA-sequencing technique and samples from cancer patients to confirm that ferroptosis tendency is positively correlated with EMT. It would be of interest to investigate the relationship between ferroptosis and EMT in a fibrotic environment. A possible mediator of ferroptosis-associated EMT is autophagy. Erastin induces autophagic flux in lung epithelial cells concomitant with elevated generation of EMT markers (N-cadherin, α-SMA, vimentin), while chloroquine, an autophagic inhibitor, attenuates EMT.[46] However, the relationship between autophagy and EMT remains unclear. The possible rationale behind autophagy-induced EMT is that ferritinophagy releases ferrous iron and subsequently triggers lysosomal leakage and further EMT.[47] Increasing evidence suggests that ferroptosis also promotes FMT. Myofibroblasts, differentiated from fibroblasts, play a vital role in IPF. Gong et al[48] reported that treating human fetal lung fibroblasts with erastin promoted FMT by increasing lipid peroxide levels and inhibiting GPX4 expression. Reinforcing this notion, Cho et al[49] demonstrated that in fibrotic regions, hepatocytes and HSCs undergo ferroptosis, and treatment of HSCs with RSL3 contributes to their activation and transformation into myofibroblast-like cells, supported by elevated level of plasminogen activator inhibitor-1 (PAI1), a marker of activated HSCs. Therefore, one would expect that the death of fibrogenic-promoting cells would alleviate fibrosis; indeed, many pharmacological modulations target ferroptosis in HSCs as anti-fibrotic therapy, which will be discussed later. Overall, the role of ferroptosis in HSC activation and fibrogenesis remains controversial.
In addition to EMT and FMT, ferroptotic cells can also directly release pro-fibrotic factors. Human PTECs that underwent ferroptosis induced by RSL3 administration or knockdown of GPX4 directly released pro-fibrotic factors, such as TGF-β, connective tissue growth factor (CTGF), and PDGF, which promoted fibroblast proliferation and fibroblast-to-myofibroblast differentiation.[24] A recent investigation into the synergistic relationship between ferroptosis and fibrosis in the context of CKD demonstrated that alterations in iron metabolism can result in ferroptosis within remnant kidneys. By exposing CKD kidney samples to the ferroptosis inducer cisplatin, researchers observed upregulation of TGF-β1/Smad3, as well as increased level of α-SMA and collagen I, while the ferroptosis inhibitor deferoxamine (DFO) alleviated this effect.[50] In Table 1, we list potential mechanisms for the involvement of ferroptosis in fibrosis.
Table 1 - Potential mechanisms for the involvement of ferroptosis in fibrosis. Ferroptosis inducers Factors/pathway Effects Ref Iron overload FGF21/HO-1 Pro-fibrotic pathway [18] Iron overload/cisplatin TGF-β/Smad Activating myofibroblasts [19,50] Iron overload IRP2/COX Impairing mitochondria, EMT [21] Iron overload IL-4/STAT6 Macrophage polarization [34] Iron overload ROS production and p53 acetylation Macrophage polarization [35] Iron overload IL-10, TGF-β Macrophage proliferation and polarization [37] RSL3 MCP-1 Macrophage recruitment [38] Erastin/RSL3 HMGB1 Macrophage activation [29] Hypoxia NLRP3 inflammasome CD1c+ DC activation [39] Erastin/inhibition of PTGS2 (Ferritinophagy, lysosomal leakage) EMT [43,44] Erastin/RSL3 – FMT [48,49] RSL3/knockdown of GPX4 TGF-β, CTGF, and PDGF Activating fibroblasts [24]CD1c: Cluster of differentiation 1c; COX: Cytochrome c oxidase; CTGF: Connective tissue growth factor; DC: Dendritic cell; EMT: Epithelial–mesenchymal transition; FGF21: Fibroblast growth factor 21; FMT: Fibroblast–myofibroblast transition; HMGB1: High mobility group box 1; HO-1: Heme oxygenase 1; IL-10: Interleukin-10; IL-4: Interleukin-4; IRP2: Iron regulatory protein 2; MCP-1: Monocyte chemoattractant protein-1; NLRP3: Nod-like receptor family pyrin domain containing 3; PDGF: Platelet-derived growth factor; PTGS2: Prostaglandin-endoperoxide synthase 2; Ref: Reference; ROS: Reactive oxygen species; STAT6: Signal transducer and activator of transcription 6; TGF-β: Transforming growth factor-beta; –: Not available.
In conclusion, ferroptosis has a significant impact on the entire fibrosis process by intensifying tissue injury, recruiting inflammatory cells, releasing pro-fibrotic factors, stimulating the activation of myofibroblasts, and facilitating EMT [Figure 1B].
Potential Targets for Ferroptosis-Related Fibrosis TherapiesFerroptosis is helpful in eliminating problematic cells, with the most researched application being the eradication of various cancer cell types.[3] The versatility of ferroptosis has recently been recognized to extend beyond its role in the regulation of cancer. Emerging evidence suggests that ferroptosis is also a target in the regulation of fibrosis. This role can primarily be attributed to the enhancement of ferroptosis in pro-fibrotic cells, such as HSCs, and the suppression of ferroptosis in various parenchymal cells. In Table 2, we list and categorize ferroptosis-related therapies for treating fibrosis based on their underlying mechanisms.
Table 2 - Potential anti-fibrotic compounds targeting ferroptosis and related mechanisms. Compounds Potential targets Cells Regulators Key findings Mechanism Ref Ferroptosis inducers Sorafenib SLC7A11 HSC HIF-1α Downregulating HIF-1α/SLC7A11 Inhibiting ROS removal [53]
留言 (0)