In vivo animal experimentsAnimal adoption
Thirty 6-week-old female SD rats, which were specific pathogen-free (SPF) grade and weighed 170–180 g, were purchased from Shanghai Jihui Laboratory Animal Care Co., Ltd. The animal production license number was SCXK (Hu) 2017-0005. The rearing temperature was 22–25 °C and humidity was 50–80%. The rats were subjected to artificial 12-hour (h)/12 h circadian light. We followed the regulations of the Institutional Animal Care and Use Committee for Animal Experiments. The animal research was approved by the Ethics Committee of the Animal Center of the Zhejiang Eyong Pharmaceutical Research and Development Center. Animal use license number SYXK (Zhe) 2021-0033. Every effort was made to alleviate animal suffering. Adaptive feeding was conducted for a week prior to the experiment.
Establishment of PCOS model and group administration
Five rats were retained as the control group and given a normal diet, and the other 25 rats were injected subcutaneously with 6 mg/100 g dehydroepiandrosterone (DHEA) for 21 consecutive days to induce PCOS according to the previous study [15]. After five randomly selected rats were euthanized for model verification, the remaining 20 PCOS rats were randomly divided into four groups: DHEA group, NC group, overexpression lncRNA SNHG12 (oe-lncRNA SNHG12) group, and silencing lncRNA SNHG12 group (si-lncRNA SNHG12) (n = 5). The oe-LncRNA SNHG12-Lentivirus was injected intrabursally into rat ovaries with a 10 µl-syringe (Gaoge, China). The recombinant lentivirus of small interference RNA targeting LncRNA SNHG12 (si- LncRNA SNHG12) and the control lentivirus were prepared and titered to 109 TU/mL (transfection unit). The needle was inserted slowly and held in place for 5 min. Each ovary was injected twice at different sites with 10 µl each time continuously for 7 days. The rats of the NC group were injected with si-RNA-NC as the negative control.
Sample collection
All rats were weighed once a week until the day of the virus injection. Two hours after the last dose, the body weight was measured, n = 5 in each group. All rats were euthanized by CO2 inhalation and fixed on the platform, and the ovarian tissue was removed and weighed. Blood and ovarian samples were then obtained from all rats.
Enzyme-linked immunosorbent assay (ELISA) measurement
The levels of follicle-stimulating hormone (FSH) (Rat, MM-0566R1), luteinizing hormone (LH) (Rat, MM-0624R1), estradiol (E2) (Rat, MM-0575R1), progesterone (P) (Rat, MM-0551R1), and testosterone (T) (Rat, MM-0577R1) in serum were measured using ELISA kits (Meimian, Jiangsu, China) following the manufacturer’s instructions, n = 5 in each group.
Hematoxylin-eosin (H&E) staining
The ovarian tissues which were were fixed with 4% paraformaldehyde, dehydrated using gradient ethanol and xylene, and then immersed in wax. Tissues were cut into slices at 5 μm and affixed to anti-peeling slides. The slices were baked at 60 °C for 12 h, dewaxed, hydrated with xylene and gradient ethanol, and stained with hematoxylin and eosin Stain (Servicebio, G1003). Finally, ethanol with low to high concentrations was added for dehydration. Vitrification was performed using xylene, and the slices were sealed. Finally, the sections were observed under a microscope (Nikon Eclipse E100).
Quantitative real time-PCR (qRT-PCR)
Pure ovarian tissue RNA was obtained by TRIzol (Sangon Biotech, B511311) extraction and transcribed into cDNA using a reverse transcription kit (Jiangsu Cowin Biotech CW2569). Primers (designed by Basic Local Alignment Search Tool (BLAST)), DEPC, cDNA, and SYBR Green (RR820A; Takara) were used to prepare the corresponding amplification products in the PCR instrument. The primer sequences used are listed in Table 1. The housekeeping gene were Human U6 and Rat U6. Fold changes in mRNA were calculated using the 2−ΔΔCT method, n = 3 in each group.
Western blot
Following the lysis of total protein of ovarian tissue, the BCA method was used to determine the protein concentration. After the loading buffer was applied, the boiling protein was denatured. Total protein was separated by electrophoresis, and the corresponding proteins were transferred to a membrane. The non-specific antigen was blocked with 5% milk, and the membrane containing the protein was incubated with the target antibodies (Affinity, USA): against Cyclin D1 (AF0931), CDK2 (AF6237), Bax (AF0120), Bcl-2 (AF6139), and the housekeeping control GAPDH (AF7021). After incubation at 4 °C for 14–16 h, unbound antibodies were washed and the membrane was additionally incubated with secondary antibodies. The unbound antibodies were washed again and captured using an ECL luminescence imager, n = 3 in each group.
In vitro cell culture and treatment
Human granulosa tumor cell lines (KGN, COV434, and SVOG) and normal ovarian surface epithelial cells IOSE80 were purchased from the Shanghai Institutes for Biological Sciences, China. cell lines were cultured in DMEM supplemented with 100 IU/mL streptomycin, 100 IU/mL penicillin, and 10% fetal bovine serum (GIBCO, Carlsbad, CA,)/F12 medium (GIBCO, Carlsbad, CA, USA) and incubated in an incubator at 37 °C with 5% CO2 and. Different concentrations of insulin (5, 10, 25, and 30 ng/mL) were used to stimulate SVOG, COV434, and KGN cells to induce PCOS. Based on the effects of insulin on GCs, the groups were further divided into insulin, insulin + siRNA-NC, and insulin + siRNA-lncRNA SNHG12 groups.
CCK-8 assay
COV434 and KGN cell suspensions were seeded in 96-well plates and incubated for 24 h. Each well was filled with 10 µL of the CCK-8 assay (MCE, HY-K0301) solution and incubated for 24, 48, 72, and 96 h. Microplate readers were used to measure absorbance at 450 nm, n = 5 in each group.
5-Ethynyl-2’-deoxyuridine (EdU) detection
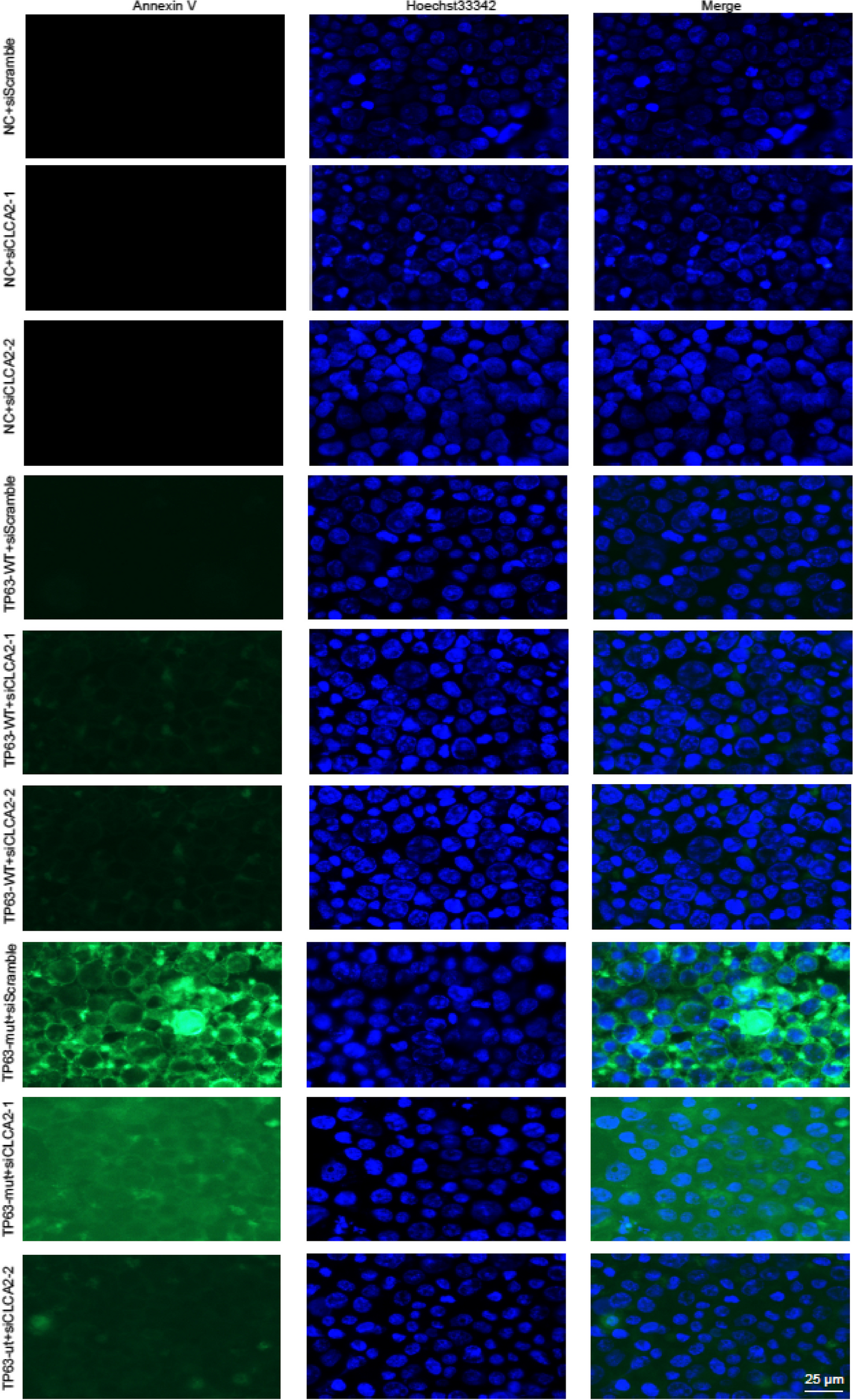
KGN cells were cultured in 12-well plates with 60–70% cell density. The Cells were exposed to EdU (Beyotime, C0078s) for 4 h at 37 °C after transfection for 48 h. The samples were fixed in 95% ethanol for 15 min at room temperature. Hoechst 33,342 solution was then applied to each well and incubated for 2 min under light, and the results were evaluated by fluorescence microscopy (Ts2-FC, Nikon, Tokyo, Japan), n = 3 in each group.
Flow cytometry (FCM)
KGN cells (1.2 × 106 per well in 6-well plates) were collected after 24 h of treatment according to the groups and washed with PBS. The supernatant was discarded by centrifugation, and 100 µL of binding buffer was added. Then, 10 µL of PI and 5 µL of Annexin V-FITC were added and thoroughly mixed. The reaction was performed in the dark for 15 min. Then, 400 µL of binding buffer was added, and the apoptosis rate was detected by FCM within 1 h, n = 3 in each group.
Quantitative real time-PCR (qRT-PCR)
Pure COV434 and KGN cell RNA was obtained by TRIzol (Sangon Biotech, B511311) extraction and transcribed into cDNA using a reverse transcription kit (Jiangsu Cowin Biotech CW2569). Primers, DEPC, cDNA, and SYBR Green (RR820A; Takara) were used to prepare the corresponding amplification products in the PCR instrument. The primer sequences used are listed in Table 1. The housekeeping gene were Human U6 and Rat U6. Fold changes in mRNA were calculated using the 2−ΔΔCT method, n = 3 in each group.
Western blot
Following the lysis of total protein of COV434 and KGN cells, the BCA method was used to determine the protein concentration. After the loading buffer was applied, the boiling protein was denatured. Total protein was separated by electrophoresis, and the corresponding proteins were transferred to a membrane. The non-specific antigen was blocked with 5% milk, and the membrane containing the protein was incubated with the target antibodies (Affinity, USA): against Cyclin D1 (AF0931), CDK2 (AF6237), Bax (AF0120), Bcl-2 (AF6139), and the housekeeping control GAPDH (AF7021). After incubation at 4 °C for 14–16 h, unbound antibodies were washed and the membrane was additionally incubated with secondary antibodies. The unbound antibodies were washed again and captured using an ECL luminescence imager, n = 3 in each group.
Dual-luciferase reporter gene assay
The luciferase reporter vector was constructed. Accordingly, the luciferase was linked to the lncRNA SNHG12 gene and then transfected into cells for detection. 24 h post-transfection, luciferase activities were analyzed in KGN cells using the dual-luciferase reporter assay (Solarbio, D0010-100T), n = 3 in each group.
RNA binding protein immunoprecipitation (RIP) assay
The Pierce RNA 3’-end Magna RIP™ RNA-Binding Protein Immunoprecipitation Kit (Sigma Aldrich, 17–700) was used to label the RNAs with biotin. Next, different groups of RNAs were cultured with cell lysates. Subsequently, the magnetic beads were added to each binding reaction at room temperature and washed. Finally, qRT-PCR was used to test the expression of miR-129 and miR‐125b, n = 5 in each group.
Statistical analysis
Statistical analysis was performed using the SPSS software (version 16.0, IBM, USA). When the measurement data in multiple groups were in accordance with a normal distribution and homogeneity of variance test, one-way ANOVA was used, and the Tukey test was used for further pairwise comparison between groups. Dunnett’s T3 test or an independent sample t-test was used if the distribution was normal, but the variance was not uniform. If it did not conform to the normal distribution, the Kruskal-Wallis H test was used. The significance level was set at P < 0.05. The data are shown as mean ± standard deviation.





留言 (0)