Enzyme I (EI) initiates the carbohydrate phosphotransferase system (PTS), a signal transduction pathway that controls the central carbon metabolism in bacteria as well as of several other cellular functions, including chemotaxis, biofilm formation, catabolic gene expression, potassium transport, and inducer exclusion[1], [2], [3], [4], [5]. This transduction pathway consists in a series of phosphoryl group transfer reactions that modulate active transport of sugars across the membrane. The transfer cascade is initiated by binding of phosphoenolpyruvate (PEP) to EI, which induces a series of intra and interdomain conformational rearrangements and results in phosphorylation of EI at His 189. The phosphoryl group is then transferred to the histidine phosphocarrier protein HPr and ultimately onto the incoming sugar[6], [7], [8], [9], [10]. EI is conserved among both Gram-positive and Gram-negative bacteria, and does not share any significant sequence similarity with eukaryotic proteins, making it an attractive target for development of wide-spectrum antimicrobials[11], [12].
EI is a 128-kDa homodimer, each subunit comprising two structurally and functionally distinct domains. The N-terminal domain (EIN, residues 1–230) is itself subdivided into two subdomains: EINα (residues 33-143) includes the binding site for the His phosphocarrier protein (HPr), and EINα/β (residues 1-20 and 148-230) contains the phosphorylation site His 189. The C-terminal domain (EIC, residues 261–575) is responsible for dimerization and contains the binding site for PEP13. Binding of PEP shifts the conformational equilibrium from an inactive dimeric open state into a catalytically competent closed state (Fig. 1A). Prior to the open-to-closed transition, EI undergoes another key large-scale conformational rearrangement in the form of a monomer–to-dimer transition6. The EI monomeric state is inactive and unable to bind PEP[14], [15]. Since both oligomeric states are present at physiological concentrations of EI (1 - 10 μM), the monomer–dimer equilibrium of the enzyme has long been suggested to be a major regulatory element for PTS[14], [16]. Therefore, EI is the prototypic oligomeric, multidomain enzyme whose activity depends on a synergistic coupling among intradomain, interdomain, and intersubunit conformational equilibria regulated by ligand binding (Fig. 1A).
We have recently reported the first atomic-level structural characterization of isolated EIC monomeric state using a combination of high-pressure NMR and molecular simulations15. Three mutations (R400E, D440R, and R559E) were introduced to disrupt salt bridges at the dimerization interface in order to overcome the high thermodynamic stability of the EIC dimeric state. The EIC variant (3m-EIC) was found to form a functional dimer at atmospheric pressure that fully dissociates into a stable monomeric state in high-pressure conditions. Pressure shifts the equilibrium toward the state that occupies the smallest partial volume. In the case of a monomer-dimer equilibrium, the monomeric state typically occupies a smaller volume because of the increased hydration of the dimerization interface that becomes accessible to solvent upon monomerization[15], [17]. Backbone residual dipolar couplings combined with accelerated molecular dynamics simulations revealed that three catalytic loops located near the dimerization interface become largely disordered upon monomerization15. This study uncovered the presence of an allosteric pathway connecting the dimerization interface and the substrate binding site, which explains why the monomeric enzyme is not able to bind its natural substrate. In particular, we found the establishment of the EIC dimer to stabilize the fold of the PEP binding site, therefore allowing formation of the enzyme-substrate complex (Fig. 1A). In another set of studies, we revealed that the EIC intradomain dynamics regulate the open-to-closed equilibrium in full-length EI. In particular, we found that the open-to-closed transition, which involves a large-scale reorientation of EIN relative to EIC, is associated with a significant reduction in interdomain flexibility, and that quenching of EIC intradomain disorder upon PEP binding is necessary to reduce the entropic cost to transition from the inactive open state to the active closed state[18], [19]. Although the studies summarized above clearly indicate an active coupling among intradomain, interdomain, and intersubunit conformational equilibria in EI (Fig. 1A), little is known about the contribution of interdomain flexibility toward the EI monomer-to-dimer transition. In particular, it is not well understood why the dimerization dissociation constant of isolated EIC is significantly lower than the dimerization dissociation constant of full-length EI20. Characterization of the structural and dynamical properties of the monomeric state of the full-length enzyme, including both EIN and EIC, is necessary to comprehensively describe the full catalytic cycle of EI.
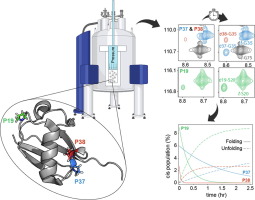
Here we use hydrostatic pressure perturbation to investigate the dimer-to-monomer transition of full-length EI. Pressure stability of wild type EI (wt-EI) was compared to that of variant 3m-EI bearing the same three salt-bridge disrupting mutations that have been recently shown to facilitate the pressure-induced dissociation of the isolated EIC dimer15. Solution NMR experiments were conducted from 1 bar to 2.5 kbar by monitoring the response to pressure of individual backbone amide and methyl groups. NMR experiments were complemented by SAXS experiments carried out in high-pressure conditions. The results consistently indicate that 3m-EI fully dissociates into a stable monomeric state above 1.5 kbar. A structural analysis of 3m-EI monomeric state was conducted by collecting backbone residual dipolar couplings (RDC) at 2 kbar. A conformational ensemble consistent with the experimental RDC data was generated using a recently described refinement protocol that takes advantage of the high computational efficiency of coarse-grained molecular dynamics (cgMD)18. The structural analysis revealed that the dimer-to-monomer transition does not result in any significant local unfolding of the EIN domain but is associated with a significant increase in interdomain flexibility that increases the entropic cost for dimer formation and explains why the isolated EIC dimer is more thermodynamically stable that the full-length EI dimer. Altogether, this study highlights the interplay between local and global conformational fluctuations in regulation of EI, and suggests that the EI catalytic cycle is coupled to a general decrease in conformational entropy, at both intra and interdomain level.






留言 (0)