
記住我


The workflow pertaining to the present study was shown in Fig. 1. The data sets related to nephroblastoma were downloaded from the TARGET database, and a total of 1839 DEGs were identified, including 1087 up-regulated genes and 752 down-regulated genes. Subsequently, the volcanic map and heat map of these DEGs were plotted (Fig. 2A, B). The expression data profiles of DEGs and their clinical data would be used for the subsequent WGCNA.
Fig. 1
The flowchart of this study
Fig. 2
Identification of differential genes and WT-related genes were screened by WGCNA. A Volcanic map of DEGs; B heat map of DEGs; C heat map analysis of correlation between modules for β = 4; D analysis of the scale-free index for various soft-threshold powers (β); E Connectivity distribution histogram at β = 4; F Cluster dendrogram of the co-expression network modules (1-TOM); G Heat map of the correlation between characteristic gene modules and different clinical data of nephroblastoma
3.2 Construction of the gene co-expression networkThe gene co-expression network was constructed with the assistance of the WGCNA package in R software. It was found that outliers were not required to be eliminated, and hence all 1904 DEGs were included in WGCNA. Given the construction of the scale-free network and the moderate retention of average connectivity, β = 4 was selected to construct the co-expression network (the correlation coefficient = 0.98 was selected as the standard), as shown in Fig. 2C and D. All DEGs were divided into 9 modules based on WGCNA (Fig. 2E, F). In order to infer the clinical relevance of these genes, these gene modules were combined with the clinical data of patients with nephroblastoma. Tumor staging and survival data of patients were important evaluation indexes for selecting functional modules. The results demonstrated that the eigenvalue of the yellow module highly correlated with the histological type and pathological stage of patients with nephroblastoma, as shown in Fig. 2G. In order to explore the molecular markers related to prognosis, these lncRNAs in this module were selected for the subsequent survival analysis.
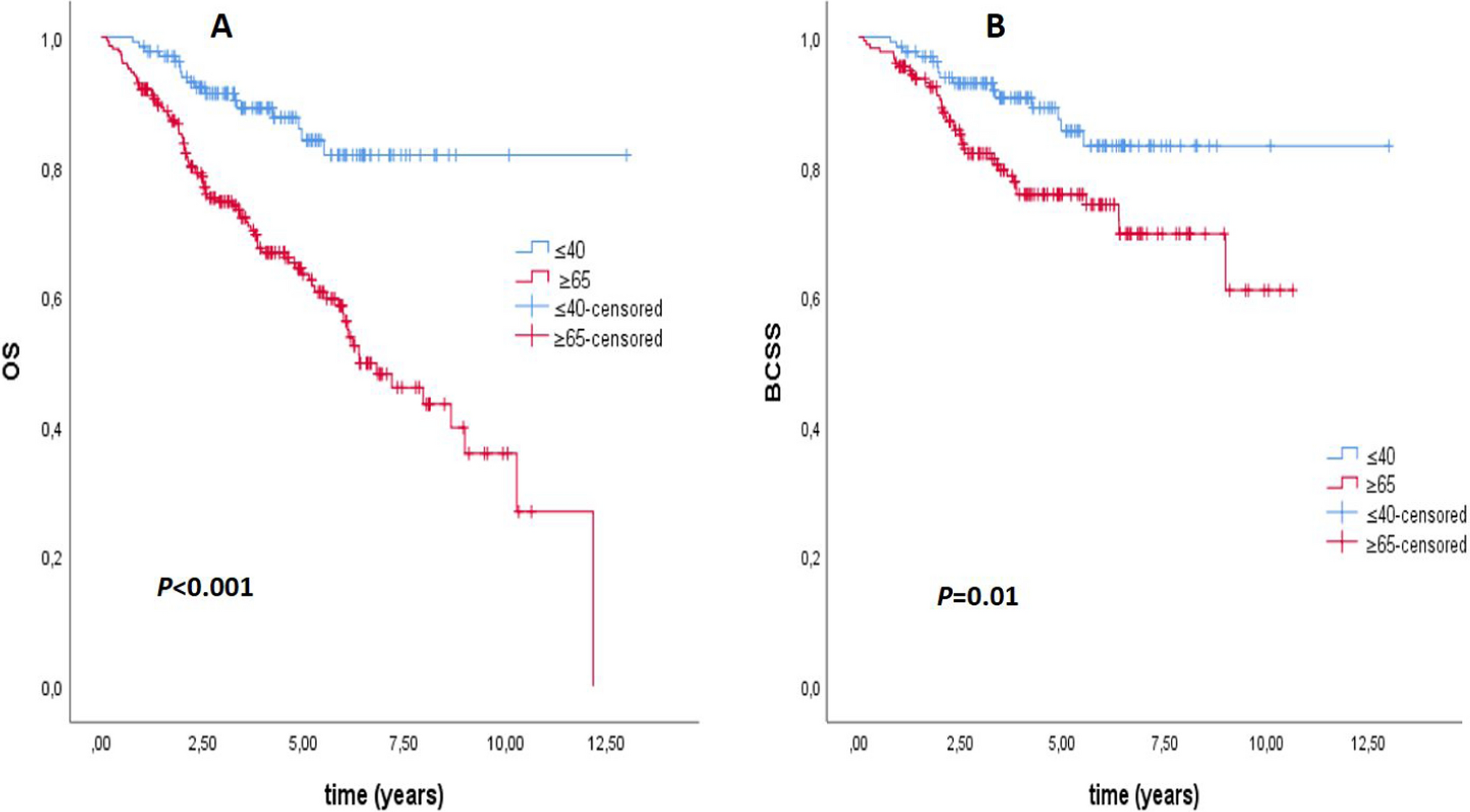
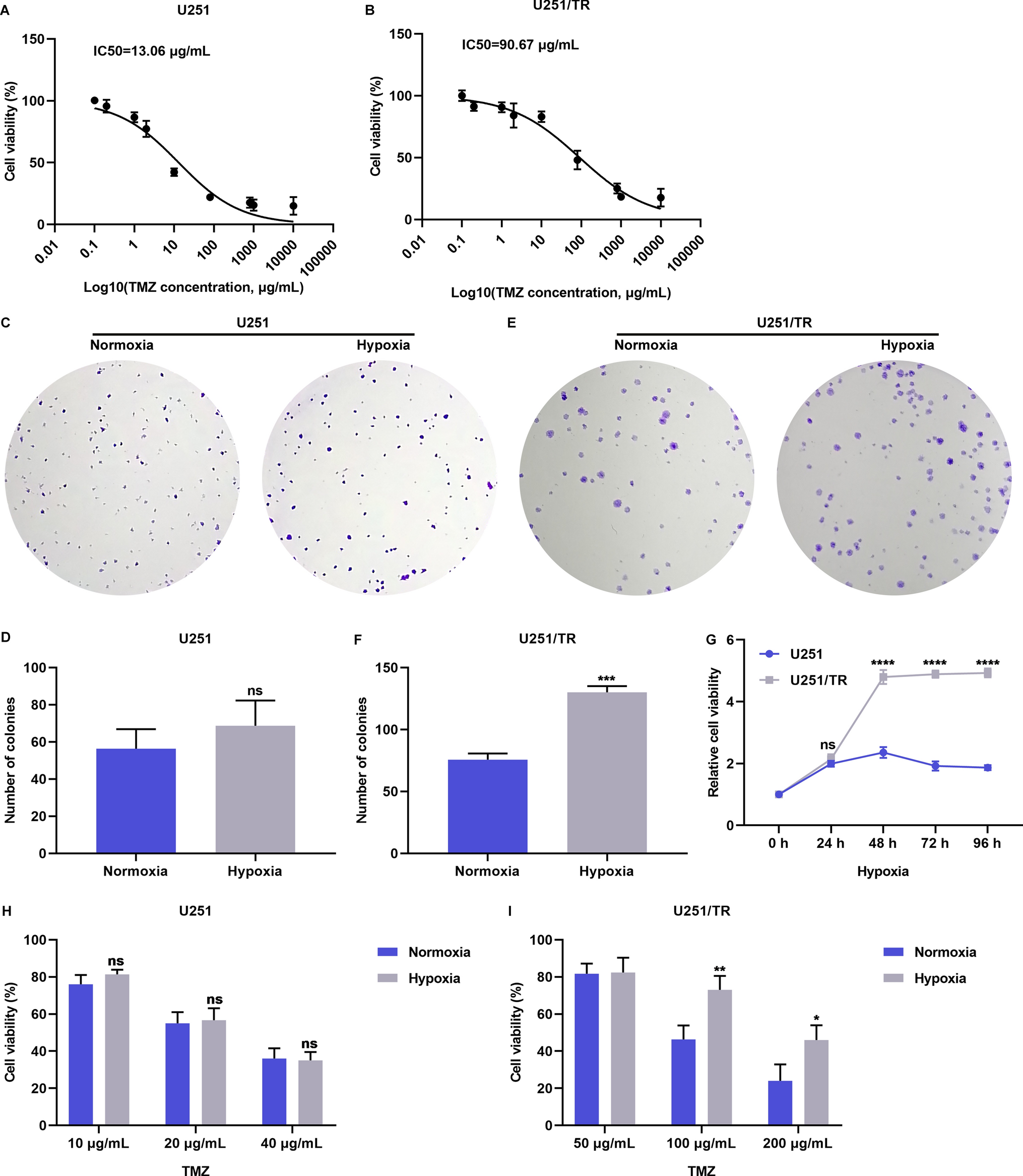
3.3 Identification of LncRNAs related to the overall survival of patients with nephroblastomaThe modules with a strong correlation with the prognosis of patients with nephroblastoma were selected. Then, the module significance (MS) of each module was calculated after each module was correlated with clinical features. The higher the MS value, the more important the module. Based on MS comparison, the modules with a strong correlation with a certain clinical feature were regarded as hub modules. Subsequently, the gene significance (GS) and module membership (MM) were calculated. In the hub modules, the genes with │MM│ > 0.8 and │GS│ > 0.2 were regarded as candidate hub genes. As a result, 11 candidate hub genes were obtained, including CTD-2006C1.2, RP4-785G19.5, SNHG1, LRP4-AS1, ZFAS1, RP11-290L1.5, FOXC2-AS1, SNHG5, PXN-AS1, CCNT2-AS1, and SNHG15. The clinical data of patients with nephroblastoma were downloaded from the TARGET database. The Kaplan–Meier survival analysis was performed on the lncRNAs of 11 candidate hub genes with the assistance of the survival package in R software. These patients with nephroblastoma were divided into two groups according to the best cut-off value of each lncRNA expression value. The Kaplan–Meier survival analysis results revealed that the overall survival rate of patients in the SNHG15 high expression group was lower (P = 0.011), as shown in Fig. 3B. The time-dependent ROC curve indicated that SNHG15 was accurate in predicting the 1-year, 2-year, and 3-year survival rates, and the area under the curve (AUC) was 0.618, 0.551, and 0.542, respectively (Fig. 3C). The univariate Cox regression analysis results suggested that SNHG15 (HR = 1.196, P = 0.008) can be regarded as an independent prognostic factor (Fig. 3A).
Fig. 3
Clinical characterization and functional enrichment analysis of SNHG15. A Univariate Cox regression analysis of Overall survival and forest plots; B Kaplan–Meier curve result. C The AUC of the prediction of 1, 2, and 3-year survival rates of WT; D Expression of SNHG15 in the validation set (GSE66405); GO function enrichment analysis bubble chart (E) and KEGG signaling pathway enrichment analysis bubble chart (F) of SNHG15-related mRNAs in the starBase data-base; G The LncACTdb3.0 database was adopted to analyze SNHG15-related ceRNAs and plot the ceRNA correlation circle diagram
3.4 Expression and function prediction of SNHG15 in nephroblastomaThe expression of SNHG15 in nephroblastoma was verified with another independent data set (GSE66405) from the GEO database (https://www.ncbi.nlm.nih.gov/). The statistical analysis results suggested that the average expression level in the normal group and the WT group was 9.012 ± 0.197 and 9.844 ± 0.555, respectively. The independent samples T test results suggested that the average expression level in the WT group was higher than that in the normal group (Fig. 3D). The difference between both groups was 0.831 (0.253–1.41). The expression level of SNHG15 in nephroblastoma was higher than that in normal kidney tissues, and there was a significant difference (t = 2.934, P = 0.006). The SNHG15-related mRNAs were predicted based on the StarBase database and LncACTdb3.0 database. Besides, the functional enrichment analysis and signal pathway enrichment analysis were carried out on the downstream target genes to infer the function of SNHG15, and the results were visualized (Fig. 3G). According to GO analysis, the down-stream target genes of SNHG15 were mainly involved in methyltransferase activity, DNA modifying enzyme, post-transcriptional regulation of gene expression, regulation of cellular amide metabolism, mRNA synthesis, and the processing, splicing and binding of RNA (Fig. 3E). According to KEGG analysis, the downstream target genes were mainly involved in splice formation, PI3K/AKT signaling pathway [29], IL-17 signaling pathway, AMPK signaling pathway, and cell cycle and apoptosis regulation, as shown in Fig. 3F. The PI3K/AKT signaling pathway is an intracellular signal transduction pathway, and it can respond to extracellular signals and promote metabolism, proliferation, cell survival, growth, and angiogenesis.
3.5 Immune infiltration characteristics of patients with high and low levels of SNHG15To reveal differences of immune infiltration in patients with high and low levels of SNHG15, we evaluated the proportions of stromal and immune cells of patients in the TARGET cohort, and found that stromal and tumor purity were higher in patients with high SNHG15 expression (Fig. 4A, B). Then, the relationship between SNHG15 and the immune component of patients in the WT group was assessed by the ESTIMATE and ssGSEA algorithms. The results indicated that there was a significant difference in the immune cells and immune function between both groups. B cells, M0 and M1 macrophages were found to be fewer in number while M2 macrophages, eosinophils, and neutrophils were more abundant in the patients with high SNHG15 expression. Moreover, the immune checkpoints were identified and there were significant differences in CD80, CD48, IDO2, CD276, CD28, and CD200R1 between both groups (Fig. 4C–E) [30].
Fig. 4
Analysis of the tumor immune microenvironment in high- and low- expression SNHG15 groups. A Heat map of the immune status of patients in both groups based on the ssGSEA algorithm; B boxplot of immune scores between both groups; C boxplot of immune checkpoint expression between both groups; D boxplot of immune cell infiltration level between both groups; E boxplot of differences in immune function between both groups (*P < 0.05; **P < 0.01; ***P < 0.001)
3.6 Identification of WT cell subtypesThe scRNA-seq data (GSE200256) used for analysis was filtered using the GEO database. We first filtered the scRNA-seq data using the R package "Seurat" for data processing, which filtered out unqualified cells for subsequent analysis (Fig. 5A, B). Then, the FindVariableFeatures function was used to find highly variable genes, and we found that 1500 genes were highly variable (Fig. 5C). The data were normalized by log-normalization. All genes were scaled using the ScaleData function and subjected to principal component analysis (PCA) downscaling, single-cell samples were scattered and distributed with logical results (Fig. 5D). Meanwhile, in PCA, we also selected 20 principal components (PCs) [31] with P.value less than 0.05 for subsequent analysis (Fig. 5E, F). Then, the core cells were classified into 19 independent cell clusters using the Uniform Manifold Approximation and Projection (UMAP) and t-distributed Stochastic Neighbor Embedding (t-SNE) algorithm (Fig. 5G, H) [32].
Fig. 5
The scRNA-seq data (GSE200256) used for analysis was filtered. A, B The scRNA-seq data were filtered to filter out ineligible cells for subsequent analysis; C the variance plot shows the variation of gene expression in all cells of WT. The red dots represent highly variable genes and the black dots represent non-variable genes; D PCA showed a clear separation of cells in WT; E, F PCA identified the top 20 PCs at P < 0.05; the core cells were classified into 19 independent cell clusters using the Uniform Manifold Approximation and Projection (UMAP) (G) and t-distributed Stochastic Neighbor Embedding (t-SNE) algorithm (H)
The different cell clusters were annotated by finding marker genes through the "singleR" package (v1.8.1), CellMarker database, and PanglaoDB database (Fig. 6A), resulting in 10 cell clusters, namely B cells, cancer cells, M2 macrophages, regulatory T cells, plasma cells, monocytes, CD8+ T cells, monocytes, fibroblasts, and epithelial cells (Fig. 6B, C) [33]. Specific gene marker expression in each cell type was shown in the form of a Heatmap (Fig. 6D).
Fig. 6
Annotation of the different cell clusters yielded 10 cell clusters. A Bubble plots of expression levels of marker genes for each cell cluster; B, C ten cell clusters were annotated based on the expression of marker genes; D Heatmap of specific gene markers in each cell type; E functional enrichment of "HALLMARK" was per-formed on cancer cells with high/low SNHG15 expression. F EMT among cell types exhibits het-erogeneity of violin plots; G. Differentially expressed genes between tumor cells with high/low SNHG15 expression; H SNHG15 exhibited heterogeneity among different cell types, with the largest proportion of tumor cells and M2 macrophages
In addition, functional enrichment of "HALLMARK" was performed on cancer cells with high/low SNHG15 expression with the use of "irGSEA" and "GSVA" in R. GSVA analysis implied that “wnt/β-catenin signaling”, “PI3K‐AKT‐MTOR signaling”, and “TGF-β signaling” were enriched in the cells with high SNHG15 expression (Fig. 6E). These results suggested that the high expression of SNHG15 in cancer cells might play a role in promoting M2 macrophage infiltration through these pathways, thus affecting prognosis.
Epithelial-mesenchymal transition (EMT) occurs normally throughout development, and dysregulation of EMT can lead to tumorigenesis [34]. EMT was shown to exhibit heterogeneity among different cell types (Fig. 6F). In this study, we found that SNHG15 exhibited heterogeneity among different cell types (Fig. 6G), with the largest proportion of tumor cells and M2 macrophages. The difference in the expression levels of tumor cells between the SNHG15 high expression group and the SNHG15 low expression group was statistically significant, and differentially expressed genes were visualized in the two groups (Fig. 6H).
3.7 Intercellular interactions in nephroblastomaPseudo-temporal analysis was performed separately for all clusters annotated in order to explore their differentiation directions with the Monocle 2 algorithm. The results showed that WT cells gradually followed 3 directions of differentiation (Fig. 7A, B). Epithelial cells divided earlier than other cells and differentiated into two branches, one of which was dominated by EMT (Fig. 7E), and the other branch gradually differentiated in multiple directions, dominated by tumor cells highly ex-pressing SNHG15 and M2 macrophages (Fig. 7D). Furthermore, we inferred intercellular communication networks to predict intercellular communication according to specific pathways and ligand receptors. The Heatmap of the number of ligand-receptor pairs showed that cellular communication occurred more frequently in M2 macrophages, B cells, tumor cells, and epithelial cells (Fig. 7C, G).
Fig. 7
Intercellular interactions in nephroblastoma. A, B Trajectory analysis of three WT cell subpopulations with different differentiation patterns; C number and strength of interactions be-tween key cells; D, E epithelial cells differentiate earlier into two branches, one of which is dominated by EMT, and the other branch gradually differentiates in multiple directions and is dominated by high SNHG15-expressing tumor cells and M2 macrophages; Bubble charts (F) and heat maps (G) visualizes the number of potential ligand-receptor pairs in key cells
Specifically, the frequency and intensity of interactions between cancer cells and M2 macrophages, and between cancer cells and epithelial cells were high (Fig. 7G). In addition, plasma cells, CD8+ T cells had relatively few interactions with other cells. The study of ligand-receptor pairs showed that the key receptor-ligand pairs between macrophages and tumor cells were mainly NRXN3-NLGN1/NRXN1-NLGN1/NRG3-ERBB4/NRG1-ERBB4 (Fig. 7F).
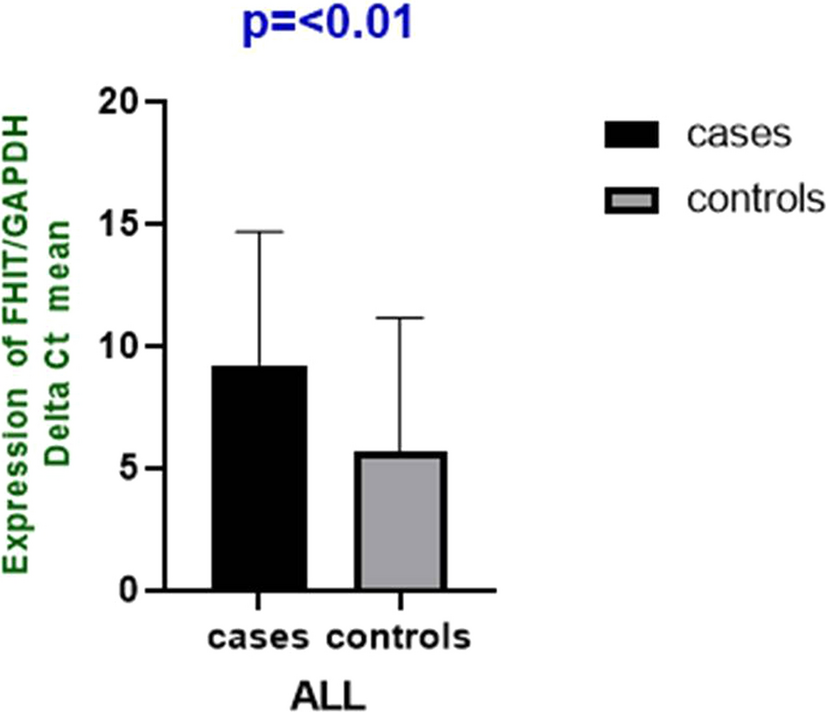
3.8 Analysis of SNHG15 pan-cancer differentially expressed and RNA-modified genesWe calculated the expression differences between normal and tumor samples in each tumor using R software and performed differential significance analysis using unpaired Wilcoxon Rank Sum and Signed Rank Tests (Fig. 8A). We observed significant upregulation in 20 tumors such as GBM (Tumor:7.39 ± 0.53, Normal:5.73 ± 1.46, p = 8.0e−63), GBMLGG (Tumor: 7.12 ± 0.52, Normal: 5.73 ± 1.46, p = 3.8e−156), LGG (Tumor:7.04 ± 0.49, Normal:5.73 ± 1.46, p = 5.1e−123), BRCA (Tumor. 7.36 ± 0.81, Normal: 7.14 ± 0.72, p = 5.9e−9), LUAD (Tumor: 7.40 ± 0.78, Normal: 7.09 ± 0.93, p = 9.9e−12), ESCA (Tumor: 7.11 ± 0.80, Normal: 6.24 ± 1.32, p = 2.0e−38), STES (Tumor: 6.97 ± 0.86, Normal: 6.09 ± 1.44, p = 1.9e−78), COAD (Tumor: 7.50 ± 0.55, Normal:6.34 ± 1.80, p = 2.8e−75), COADREAD (Tumor: 7.53 ± 0.56. Normal: 6.37 ± 1.78, p = 1.6e−87), STAD (Tumor: 6.90 ± 0.87, Normal: 5.59 ± 1.68, p = 9.8e−48), LIHC (Tumor:5.80 ± 0.90, Normal: 4.99 ± 0.56, p = 2.1e−27), WT (Tumor: 7.16 ± 0.86, Normal: 6.43 ± 1.55, p = 5.9e−9), SKCM (Tumor:7.69 ± 1.16, Normal: 6.49 ± 0.38, p = 1.5e−31), THCA (Tumor:7.49 ± 0.78, Normal:6.99 ± 1.00. p = 6.9e−33), READ (Tumor: 7.64 ± 0.58, Normal: 7.10 ± 0.47, p = 2.0e−3), PAAD (Tumor:7.19 ± 0.68, Normal: 4.76 ± 1.71, p = 1.9e−53), TGCT (Tumor: 6.35 ± 1.10. Normal: 5.75 ± 0.47, p = 3.0e−13), ALL (Tumor: 5.34 ± 1.10, Normal: 3.66 ± 1.34, p = 1.3e−29), LAML (Tumor:6.79 ± 0.62, Normal: 3.66 ± 1.34, p = 5.3e−74), CHOL (Tumor:6.10 ± 0.94, Normal: 5.22 ± 0.25,p = 2.8e−3), we observed significant downregulation in seven tumors such as UCEC (Tumor: 6.45 ± 0.99, Normal: 7.04 ± 0.46, p = 5.7e−3), CESC(Tumor:6.56 ± 0.89. Normal: 7.33 ± 0.56, p = 1.2e−3), KIRP (Tumor: 6.03 ± 0.85, Normal: 6.43 ± 1.55, p = 1.8e−12), KIPAN (Tumor: 6.40 ± 0.94, Normal: 6.43 ± 1.55, p = 0.01), OV (Tumor: 6.44 ± 1.18, Normal: 7.15 ± 0.42, p = 1.2e−13), UCS (Tumor: 6.70 ± 0.93, Normal: 7.18 ± 0.39, p = 4.8e−3), KICH (Tumor: 5.68 ± 0.92, Normal: 6.43 ± 1.55, p = 1.8e−12). SNHG15 is closely linked to marker genes for three classes of RNA modifications (m1A (10), m5C (13), and m6A (21)) genes, and the exact mechanism needs to be further explored, which confirms that SNHG15 plays a major role in tumors (Fig. 8D).
Fig. 8
Validation of SNHG15 from a pan-cancer and experimental perspective. A Analysis of SNHG15 expression in pan-cancer; B the expression of SNHG15 was measured by qRT-PCR in WT tissues and paired normal tissues; C the siRNA interference of SNHG15 significantly inhibited the expression of SNHG15 compared with the control group; D SNHG15 is closely linked to marker genes for three classes of RNA modifications (m1A, m5C, and m6A) genes; E transwell assay, siSNHG15 reduced the invasion and migration of nephroblastoma cells; F study of the effect of SNHG15 on the proliferation and viability of nephroblastoma cells using CCK-8 assay; G wound healing assay: siRNA interference with SNHG15 expression significantly inhibits the proliferation and migration of G401 cells; H the expression of EMT-related core proteins was examined by western blotting
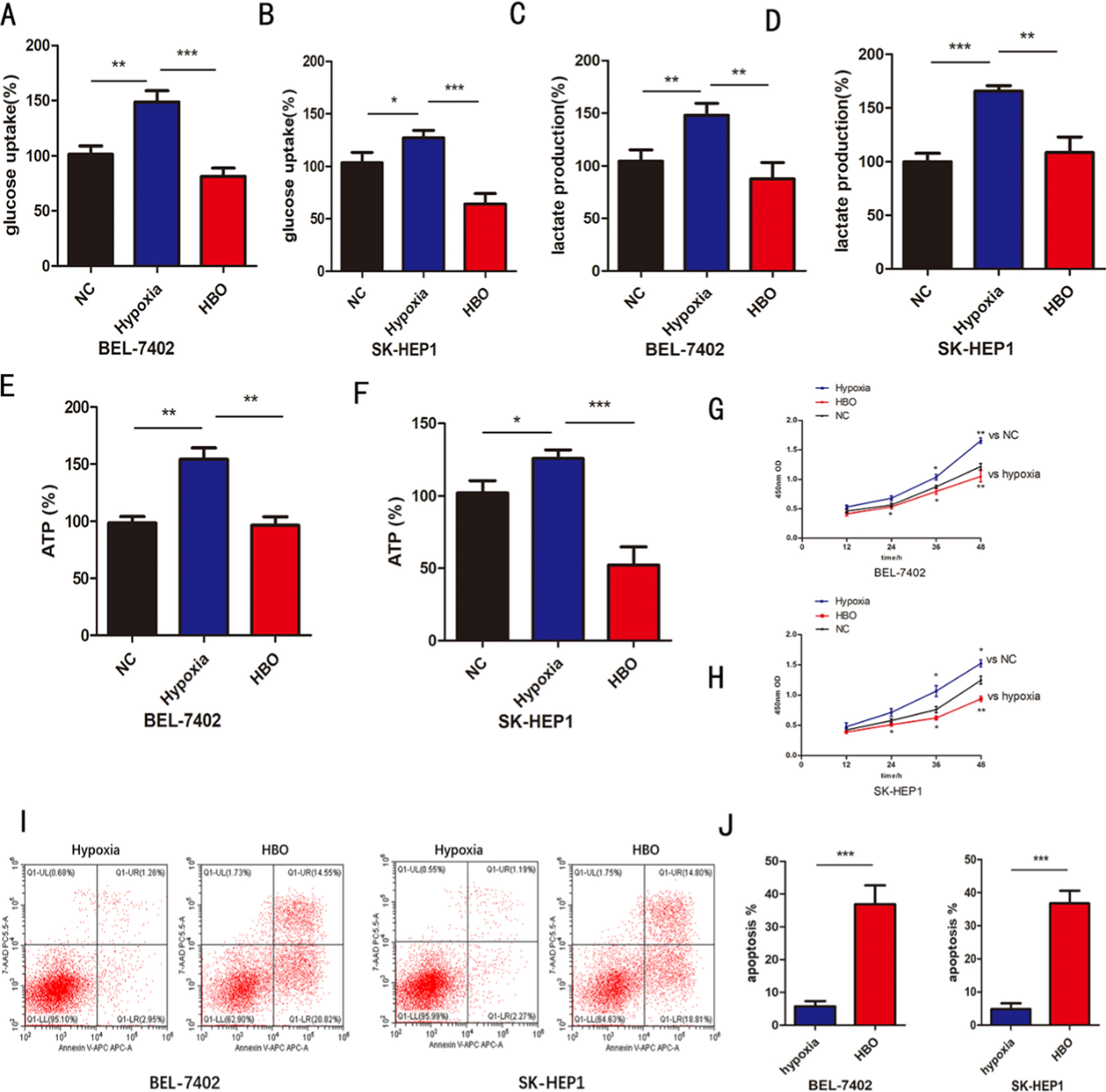
3.9 In vitro cell experiment resultsThe expression of SNHG15 was measured by qRT-PCR in WT tissues and paired normal tissues (Fig. 8B). The siRNA interference of SNHG15 significantly inhibited the expression of SNHG15 compared with the control group (P < 0.05), as shown in Fig. 8C.
In addition, a series of experiments were performed to investigate the effects of SNHG15 on the proliferation, invasion and migration of nephroblastoma cells. We investigated the effect of SNHG15 on the proliferation and viability of nephroblastoma cells using CCK-8 assay (Fig. 8F). Wound healing assay showed that siRNA interference with SNHG15 expression significantly inhibited G401 cell proliferation migration and promoted apoptosis compared with control (Fig. 8G). These results were further supported by Transwell assay, siSNHG15 reduced the invasion and migration of nephroblastoma cells, as shown in Fig. 8E. In addition, we also detected the knock-down of SNHG15 by Western blotting for the expression of EMT-related core proteins. Compared with the siNC group, knockdown of SNHG15 decreased the expression of EMT-associated core protein (Fig. 8H). According to the results of cell cycle analysis, knocking down the expression of SNHG15 resulted in its blockage in the S phase, which in turn inhibited the proliferation of tumor cells (Supplementary figure 1).
留言 (0)