
記住我

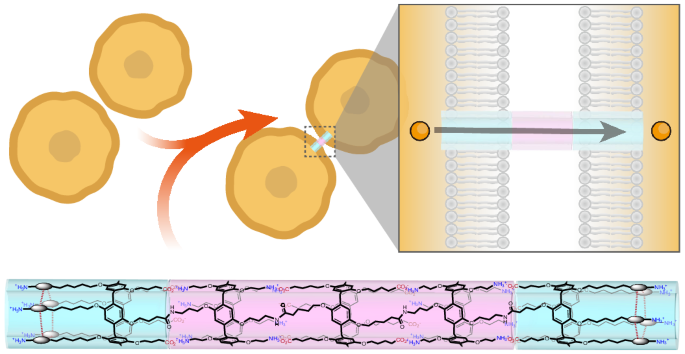

The electron donors shown in Fig. 1c were chosen to explore the influence of the following factors on cage escape: (1) the variation in the driving force for photoinduced electron transfer (ΔGET), (2) the effect of donor size (TAA-OMe, TAA-PEG3 and TAA-PEG7), (3) the influence of heavy atoms (TAA-Cl, TAA-Br and TAA-I), (4) structural differences in the aromatic amines (triarylamines (TAAs) versus N,N-dimethylanilines) and (5) aromatic amines versus aliphatic amines (2-phenyl-1,2,3,4-tetrahydroisoquinoline (THIQ), triethylamine (TEA) and N,N-diisopropylethylamine (DIPEA)).
[Ru(bpz)3]2+ and [Cr(dqp)2]3+ have similar excited-state reduction potentials (1.45 and 1.26 V versus the saturated calomel electrode, respectively)30. All 12 donors quenched the luminescent excited states of the two complexes with rate constants (kq) in the range of 108–1010 M−1 s−1 (Supplementary Figs. 15–40), revealing very similar driving-force dependence in both cases (Fig. 2c), in line with previous studies30,31,32. Deviations from this Rehm–Weller behaviour were observed for the halide-substituted TAA electron donors 4–6 (highlighted in the ellipsoids in Fig. 2c). Their heavy atoms accelerate photoinduced electron transfer22, despite the decreasing driving force (−ΔGET) along the series TAA-Cl > TAA-Br > TAA-I (Supplementary Section 7.13.3). The driving forces for the photoinduced electron transfer and in-cage thermal charge recombination for irreversible donors were estimated according to the oxidation peak potentials because standard redox potentials were not available in these cases.
Fig. 2: Cage escape quantum yields and excited-state quenching kinetics.
a, Transient absorption decays of the reference complex [Ru(bpy)3]2+ (12 µM in aerated H2O) at 455 nm and the [Ru(bpz)3]2+ (12 µM)–TAA-OMe (2 mM) pair in aerated CH3CN at 293 K at 717 nm under 422 nm excitation. Both solutions have identical absorbance at 422 nm (Supplementary Fig. 55a). The concentrations of the 3MLCT-excited [Ru(bpy)3]2+ and TAA-OMe·+ formed in this experiment were 3.14 and 1.82 ± 0.06 μM, respectively. b, Transient absorption decays of the reference complex [Ru(bpy)3]2+ (12 µM in aerated H2O) at 455 nm and the [Cr(dqp)2]3+ (30 µM)–TAA-OMe (2 mM) pair in aerated CH3CN at 293 K at 717 nm under 416 nm excitation. Both solutions have identical absorbance at 416 nm (Supplementary Fig. 56a). The concentrations of the 3MLCT-excited [Ru(bpy)3]2+ and TAA-OMe·+ were 1.56 and 0.20 ± 0.02 μM, respectively. In a and b, the relative concentrations of the photoproducts were derived from the measured ΔOD values at 455 nm for the reference (3MLCT-excited [Ru(bpy)3]2+) and at 717 nm for the donor–acceptor pairs (TAA-OMe·+), as well as the respective Δε values at the relevant observation wavelengths (Δε455 = −10,100 M−1 cm−1 for [Ru(bpy)3]2+ (ref. 33) and Δε717 = 32,600 ± 300 M−1 cm−1 for TAA-OMe·+). This analysis yielded ФCE values of 58 ± 2% for the [Ru(bpz)3]2+–TAA-OMe pair and 13 ± 1% for the [Cr(dqp)2]3+–TAA-OMe pair in CH3CN at room temperature, as marked by the dashed horizontal lines. c, Quenching rate constants (kq) for photoinduced electron transfer from the individual donors 1–12 to [Ru(bpz)3]2+ and [Cr(dqp)2]3+ plotted as a function of the reaction free energy (ΔGET). The halide-substituted donors are marked by the ellipsoids. d, Cage escape quantum yields (ФCE) obtained with [Ru(bpz)3]2+ and [Cr(dqp)2]3+ for the electron donors 1–12.
Trends in cage escape quantum yieldsThe cage escape quantum yields for the electron transfer photoproducts formed after the excitation of [Ru(bpz)3]2+ or [Cr(dqp)2]3+ in the presence of the donors 1–12 (Fig. 1c) in aerated acetonitrile were determined by relative actinometry using laser flash photolysis (Supplementary Sections 6 and 7). The basic principle of this method is to compare the concentrations of the photoproducts formed after pulsed excitation of a sample and a reference solution. The reference, here an aqueous aerated solution of [Ru(bpy)3]Cl2 (bpy, 2,2′-bipyridine), has a known quantum yield for the formation of its photoproduct(s) and serves to determine the number of photons absorbed by the reference. By adjusting the photocatalyst concentration in the main sample and in the reference solution, an equal absorbance at the excitation wavelength and an equal concentration of the excited photocatalysts are ensured for both solutions (Supplementary Section 7). Measurement of the concentration of the photoproducts formed in the main sample then permits the determination of the unknown quantum yield.
The excitation of [Ru(bpy)3]2+ at 422 nm causes a depletion of its diagnostic singlet metal-to-ligand charge transfer (1MLCT) ground-state absorption band, manifesting in a known change in its molar extinction coefficient (Δε455 = −10,100 M−1 cm−1 at 455 nm, ref. 33). This bleaching reflects the quantitative formation of a 3MLCT excited state, which decays on a microsecond timescale (Fig. 2a,b). For ease of comparison with the main samples under test, the experimentally observed changes in the optical density (ΔOD) of this reference at 455 nm were normalized to an initial value of 1.0. Concentration-adjusted samples containing the [Ru(bpz)3]2+–TAA-OMe and [Cr(dqp)2]3+–TAA-OMe donor–acceptor pairs were then excited under identical conditions. The donors were present in large excess relative to the metal complexes (millimolar versus micromolar concentrations) to ensure rapid and maximally efficient (quantitative) photoinduced electron transfer, yet selective excitation of the metal complexes remained possible in all cases due to the negligible extinction coefficient of the donors at the relevant wavelengths. The TAA-OMe·+ oxidation product was readily detected by its characteristic absorption in the red spectral range with a change in the molar extinction coefficient (Δε717) of 32,600 ± 300 M−1 cm−1 at 717 nm (Supplementary Fig. 44). The transient absorption decay curves for the two donor–acceptor pairs are shown in Fig. 2a,b, respectively. Cage escape quantum yields (ФCE) of 58 ± 2% for the [Ru(bpz)3]2+–TAA-OMe pair and 13 ± 1% for the [Cr(dqp)2]3+–TAA-OMe pair were determined from the relative intensities reached immediately after excitation. The decays of the respective transients reflect reverse electron transfer from the reduced metal complexes to TAA-OMe·+ after successful cage escape.
The same method was applied to all of the donor–acceptor pairs, which required the measurement of the extinction-calibrated difference spectra for the oxidized and neutral forms of the individual donors, as well as for the native and reduced forms of the two metal complexes (Supplementary Sections 4, 6 and 7). The resulting ФCE values obtained with [Ru(bpz)3]2+ and [Cr(dqp)2]3+ for the 12 electron donors are presented in Fig. 2d. All of the TAA-based donors underwent reversible electron transfer, whereas the other electron donors were irreversible donors due to deprotonation after the initial one-electron oxidation34. In principle, such proton-coupled electron transfer reactions could artificially enhance the cage escape quantum yields obtained for the two photosensitizers with the non-TAA donors, potentially complicating the direct comparison between TAA and non-TAA donors.
The following findings emerge from these experiments. (1) For all of the electron donors explored here, the ФCE values obtained with [Ru(bpz)3]2+ are substantially higher than those obtained with [Cr(dqp)2]3+. (2) There is no clear evidence of a heavy-atom effect on cage escape (Supplementary Section 7.13.3). (3) Increasing donor size in the series TAA-OMe < TAA-PEG3 < TAA-PEG7 leads to increasing ФCE values, which could be due to better distancing of the donor–acceptor pairs (Supplementary Section 7.13.4)26. (4) No substantial change in cage escape quantum yield was observed on variation of the electron donor concentration (Supplementary Fig. 71). (5) The presence of oxygen does not affect the cage escape quantum yields (Supplementary Figs. 55–57).
Different cage escape for Ru and Cr complexes[Ru(bpz)3]2+ and [Cr(dqp)2]3+ have fundamentally different photoactive excited states (Fig. 1b) as a result of their different valence electron configurations (low-spin d6 versus d3)30,35. The 3MLCT excited state of [Ru(bpz)3]2+ is predisposed to charge transfer, yet the 2E and 2T1 spin-flip excited states of [Cr(dqp)2]3+ promote photoinduced electron transfer similarly well according to the data in Fig. 2c. However, due to substantial in-cage charge recombination (Fig. 1a), much lower cage escape quantum yields were systematically obtained with the CrIII complex compared with the RuII complex (Fig. 2d). At first glance, therefore, it seems plausible that the differences in the electronic structures of [Ru(bpz)3]2+ and [Cr(dqp)2]3+ are the cause of the very different cage escape quantum yields. We initially considered different spin-correlated radical pairs emerging from the 3MLCT and 2E/2T1 excited states and spin effects for in-cage charge recombination as a possible reason for the systematic differences between the RuII and CrIII complexes (Supplementary Section 7.13.2), but the data currently available do not permit corroboration of this hypothesis. Instead, it seems that Marcus theory is able to account for the observed behaviour, making superfluous the involvement of spin and other effects as possible explanations. The cage escape quantum yield (ФCE) for both complexes is governed by the relative magnitudes of the cage escape rate (kCE) and in-cage reverse electron transfer (krET; equation (1a,b)).
$$_}}\left(}\right)=\frac_}}\left(}\right)}_}}\left(}\right)+_}}\left(}\right)}$$
(1a)
$$_}}\left(}\right)=\frac_}}\left(}\right)}_}}\left(}\right)+_}}\left(}\right)}$$
(1b)
Assuming equal cage escape rate constants for the RuII and CrIII complexes (kCE(Ru) = kCE(Cr)), the ratio of rate constants for in-cage reverse electron transfer can be formulated as shown in equation (2):
$$\frac_},\exp .}\left(}\right)}_},\exp .}\left(}\right)}=\frac_}}\left(}\right)}-1}_}}\left(}\right)}-1}$$
(2)
Thus, under the assumption that PC·− and D·+ escape the solvent cage equally quickly regardless of whether PC·− corresponds to [Ru(bpz)3]+ or [Cr(dqp)2]2+, the experimentally determined cage escape quantum yields can be used to estimate the ratio of in-cage reverse electron transfer for the RuII and CrIII complexes for any given electron donor D (equation (2)). Based on the data shown in Fig. 2d, the experimentally determined ratios krET,exp.(Ru)/krET,exp.(Cr) vary between 0.015 and 0.21 (Supplementary Table 4).
Within the framework of semi-classical Marcus theory (equation (3)), the rate constants for (in-cage) reverse electron transfer (krET,calc.) can be expressed as a function of the electronic coupling (HAB) between the reduced photocatalyst (PC·−) and the oxidized donor (D·+), the reaction free energy (ΔGrET) for reverse electron transfer from PC·− to D·+ and the reorganization energy (λ) accompanying that reaction. Equation (3) describes the ratio of the reverse electron transfer rate constants for the RuII and CrIII complexes, where the individual parameters (HAB, ΔGrET and λ) can be different depending on the photocatalyst–donor pair considered.
$$\frac_},}.}\left(}\right)}_},}.}\left(}\right)}=\frac_}}(})\right]}^}\exp \left[-\frac_}}\left(}\right)+\lambda \right)}^}_}}}}\right]}_}}(})\right]}^}\exp \left[-\frac_}}\left(}\right)+\lambda \right)}^}_}}}}\right]}$$
(3)
All of the relevant ΔGrET values are known (Supplementary Table 1), but the HAB and λ values are unknown. Equation (3) can be used to determine what combinations of HAB(Ru)/HAB(Cr) and λ make the ratio of calculated (in-cage) reverse electron transfer rate constants krET,calc.(Ru)/krET,calc.(Cr) equal to the corresponding experimental ratio determined with equation (2). The reorganization energy for in-cage reverse electron transfer depends on the individual photocatalyst–donor pair; however, in our analysis, the reorganization energy with different donors was allowed to vary, but for a given electron donor, identical λ values were assumed for both the RuII and CrIII complexes. Varying the HAB(Ru)/HAB(Cr) ratio between 0.3 and 1.6 (in increments of 0.1) led to the distribution of λ values shown in Fig. 3a (Supplementary Table 3). For HAB(Ru)/HAB(Cr) ratios above 0.8, plausible λ values of ~1 eV account for the experimentally observed differences in the cage escape quantum yields. Figure 3b shows the distribution of λ values collected in Fig. 3a for the individual donors. The key finding is that for any given HAB(Ru)/HAB(Cr) ratio between 0.3 and 1.6, a sizeable fraction (79–86%) of the calculated λ values fall within a range of ±15% of the median reorganization energy obtained for a given donor (Supplementary Table 3). Analogous calculations were performed by considering a plausible range of λ values and solving for the HAB ratio (Supplementary Fig. 83b). Broader screening of combinations of the HAB(Ru)/HAB(Cr) ratios (in increments of 0.1 between 0.3 and 1.6) and λ values (in increments of 0.1 eV between 0.4 and 1.6 eV) revealed that 63 out of 2,184 combinations are able to emulate the experimentally derived krET,exp.(Ru)/krET,exp.(Cr) ratios with a deviation of 15% or less (Supplementary Tables 4 and 5 and Supplementary Fig. 84). Figure 3c shows the results of screening in the same range as the experimentally obtained krET,exp.(Ru)/krET,exp.(Cr) ratios, showing the correlation between the calculated and experimental ratios of the rate constants for in-cage reverse electron transfer for 63 of the combinations.
Fig. 3: Screening of in-cage reverse electron transfer parameters.
a, Reorganization energies for given ratios of electronic couplings able to produce krET,calc.(Ru)/krET,calc.(Cr) ratios (equation (3)) in line with the experimental krET,exp.(Ru)/krET,exp.(Cr) ratios derived from equation (2). The distribution here is shown for specific coupling ratios over the applied donors. b, Reorganization energies found for the individual donors based on the analysis in a (Supplementary Table 3). The distribution here is shown for specific donors over the considered coupling ratios. In a, the data size (n) for each distribution is 12 (for 12 applied donors) and in b, the data size is 14 (HAB ratios from 0.3 to 1.6 eV in increments of 0.1 eV). The grey boxes in a and b contain the middle 50% of data, the so-called interquartile range (IQR). The whisker boundaries are drawn at the most extreme data point that is no more than ±1.5IQR from the top and bottom edges of the box53. The horizontal black lines in the boxes mark the median values, and the open squares represent the average values. c, Correlation between the calculated and experimental ratios of the rate constants for in-cage reverse electron transfer. In total, 2,184 combinations of HAB(Ru)/HAB(Cr) and λ (Supplementary Table 5) were considered (grey squares, right-hand y axis), of which 63 (Supplementary Table 4) provided agreement between calculation and experiment within a deviation of 15% (black circles, left-hand y axis). The numbers shown in c correspond to the donors 1–12. d, Generic Marcus parabola illustrating the dependence of the rate constant for in-cage reverse electron transfer (krET) on its driving force (ΔGrET). For a given electron donor, in-cage reverse electron transfer with the reduced RuII complex (black square) occurs more deeply in the inverted regime and thus is slower than in-cage reverse electron transfer with the reduced CrIII complex (black circle).
The modulations of the HAB ratios and λ values were to some extent arbitrary, and this analysis was unable to provide unambiguous insights into trends in the electronic couplings and reorganization energies. This is because the identified HAB(Ru)/HAB(Cr) and λ combinations were not necessarily mathematically unique solutions, and because other factors could vary, for example, the cage escape rate constants kCE(Ru) and kCE(Cr) in equation (1a,b). Nonetheless, some of the differences in the reorganization energies calculated for the various donors studied seem to make chemical sense. For example, the positive charge in the radical cation forms of the aliphatic donors 10–12 is probably not delocalized much further than the nitrogen and proximal carbon atoms, leading to relatively small ionic radii and large reorganization energies, whereas the cationic charge in the TAAs 1–6 and anilines 7–9 is delocalized into the aromatic rings, increasing the ionic radius and resulting in smaller reorganization energies (Fig. 3b)26. The increase in λ along the series 1–3 could be due to an increase in the degree of solvation36. The quintessential point of this analysis is that reasonable values for HAB and λ can be fed into semi-classical Marcus theory and quantitatively explain the difference between the cage escape quantum yields observed with [Ru(bpz)3]2+ and [Cr(dqp)2]3+. Other possible explanations for the different cage escape quantum yields, while not fully discarded at this point, are therefore currently pushed into the background. Within this overall framework, for any given electron donor, in-cage reverse electron transfer with the RuII complex occurs ~0.3 eV more deeply in the Marcus inverted regime than with the CrIII complex, making in-cage charge recombination slower and cage escape quantum yields higher for the RuII complex (Fig. 3d).
Correlation of photocatalytic performance with cage escapeAs [Ru(bpz)3]2+ and [Cr(dqp)2]3+ show similar photoinduced electron transfer behaviour (Fig. 2c) but systematically different cage escape quantum yields (Fig. 2d), using these two photocatalysts permitted investigation of how photoredox reactions are affected by cage escape. To address this fundamental issue, three different photochemical reactions involving three distinct electron donors were investigated: (1) the photocatalytic aerobic hydroxylation of 4-methoxyphenylboronic acid using DIPEA (Fig. 4a and Supplementary Section 8.1), (2) the photocatalytic reductive debromination of 2-bromoacetophenone with N,N-dimethyl-p-toluidine (DMT; Fig. 4d and Supplementary Section 8.2) and (3) a photocatalytic aza-Henry reaction, where THIQ acts both as the electron donor and substrate (Fig. 4g and Supplementary Section 8.3). Each reaction was performed with both [Ru(bpz)3]2+ and [Cr(dqp)2]3+ under identical conditions to enable comparison (Supplementary Section 8). The concentrations of the photocatalysts were adjusted to ensure the absorption of the same amount of light emitted by the 415 nm light-emitting diode (LED) and thus the same concentration of the excited-state photocatalysts (*PC), considering the quantitative intersystem crossing for both complexes (Supplementary Section 7 and Supplementary Figs. 90, 100 and 109). This LED emitted a power density of 73 mW cm−2 at the sample position, which permitted monitoring of the reaction kinetics and minimized photodecomposition. The reaction set-up furthermore allowed the determination of the quantum yields for the formation of the photoproducts (ФP) as a function of irradiation time (Supplementary Section 9).
Fig. 4: Photoredox reactions performed with Ru and Cr catalysts.
a,d,g, Aerobic hydroxylation of 4-methoxyphenylboronic acid (a), reductive debromination of 2-bromoacetophenone (d) and aza-Henry reaction (g). The concentrations of [Ru(bpz)3]2+ and [Cr(dqp)2]3+ were adjusted to ensure equal absorbance at the irradiation wavelength of 415 nm. b,e,h, The product yields (η) of 4-methoxyphenol (b), acetophenone (e) and THIQ-MeNO2 (h) as a function of irradiation time for [Ru(bpz)3]2+ (green circles) and [Cr(dqp)2]3+ (blue triangles). Linear regression fits (orange lines) to the data points over the initial irradiation period provide the initial product formation rates ν(RuII) and ν(CrIII). c,f,i, Cage escape quantum yields (ФCE) obtained with the [Ru(bpy)3]2+ reference (dark-grey traces), [Ru(bpz)3]2+ (green traces) and [Cr(dqp)2]3+ (dark-blue traces) and the electron donors DIPEA (10 mM) (c), DMT (10 mM) (f) and THIQ (2 mM) (i), used in the photoredox reactions in a, d and g, respectively. For donor THIQ, only an upper limit could be determined for ФCE with [Cr(dqp)2]3+ (Supplementary Section 7.10). j, Experimentally determined ratios of the cage escape quantum yield (ФCE), the initial product formation rate (ν) and the initial product formation quantum yield (ФP) for [Ru(bpz)3]2+ and [Cr(dqp)2]3+ in the three photochemical reactions in a, d and g using DIPEA, DMT and THIQ, respectively, as electron donors.
The photocatalytic aerobic hydroxylation of phenylboronic acids (Fig. 4a) is well known and therefore was identified as a relevant benchmark reaction37,38. Its mechanism involves an initial electron transfer between the excited photocatalyst and an electron donor (Supplementary Fig. 89), for which DIPEA was a suitable choice. Both [Ru(bpz)3]2+ and [Cr(dqp)2]3+ photocatalysed the reaction of 4-methoxyphenylboronic acid to 4-methoxyphenol, but at very different rates (Fig. 4b), despite the identical amount of light absorbed. Based on 1H NMR experiments (Supplementary Section 8.1), unwanted side products were not formed in substantial quantities and the initial product formation rates (v) were determined to be 232 ± 4 mM h−1 with [Cr(dqp)2]3+ and 848 ± 77 mM h−1 with [Ru(bpz)3]2+, respectively (Fig. 4b), corresponding to a ratio of ν(RuII)/ν(CrIII) of 3.7 ± 0.3. For the initial product formation quantum yields, a ratio of ФP(RuII)/ФP(CrIII) of 3.3 ± 0.4 (Supplementary Fig. 117) was obtained. These ratios come close to the cage escape quantum yield ratio of ФCE(RuII)/ФCE(CrIII) = 4.0 ± 0.7 determined for the [Ru(bpz)3]2+–DIPEA and [Cr(dqp)2]3+–DIPEA couples (Fig. 4c,j). Under the photocatalytic reaction conditions, the difference between kq for the RuII and CrIII complexes was unimportant because excited-state quenching by photoinduced electron transfer was essentially quantitative in the presence of 0.25 M DIPEA (Supplementary Section 8.1).
The debromination of α-bromoketones is another well-explored benchmark photoredox reaction39,40; we chose to perform this reaction with a different electron donor (DMT) that should result in a bigger difference in the cage escape quantum yields for [Ru(bpz)3]2+ and [Cr(dqp)2]3+ (Fig. 2d). Under the conditions used for the reductive debromination of 2-bromoacetophenone (Fig. 4d), 99% of the 415 nm excitation light was absorbed by the [Ru(bpz)3]2+ and [Cr(dqp)2]3+ photocatalysts, and any background reactivity caused by the direct absorption of light by the substrate was negligible (Supplementary Section 8.2). Analogously to the aerobic hydroxylation reaction, the initial step is a photoinduced electron transfer from the electron donor to the photocatalyst (Supplementary Fig. 99)41, which, in the presence of 0.1 M DMT, is essentially quantitative and therefore not performance limiting for either photocatalyst (Supplementary Section 8.2). Several side products, including a pinacol coupling product42 and a DMT dimer, were formed in minor amounts (Supplementary Figs. 99, 101 and 102), but the anticipated formation of acetophenone clearly dominated. The initial product formation rates were determined to be 2.07 ± 0.06 mM h−1 with [Ru(bpz)3]2+ and 0.24 ± 0.01 mM h−1 with [Cr(dqp)2]3+ (Fig. 4e), corresponding to a ν(RuII)/ν(CrIII) ratio of 8.6 ± 0.5. The ratio of the initial product formation quantum yields ФP(RuII)/ФP(CrIII) was 8.4 ± 0.9 (Supplementary Fig. 118). Both of these ratios come close to the ratio of the cage escape quantum yields, with ФCE(RuII)/ФCE(CrIII) = 10.6 ± 1.3 for the DMT donor used here (Fig. 4f,j). After successful cage escape, there was no measurable difference in the onward reaction rates of the RuII and CrIII complexes within the limits of the experiments performed (Supplementary Section 8.2 and Supplementary Fig. 107).
As a third example, we chose the visible light-mediated aza-Henry reaction of nitromethane with THIQ (Fig. 4g), which is a frequently explored photocatalytic process involving C–H activation43,44. This reaction is known to occur by both the excitation of a photocatalyst and direct excitation of THIQ (Supplementary Fig. 108)43. Control experiments performed in the absence of photocatalyst confirmed this earlier finding (Supplementary Fig. 112) and signalled the formation of a known hydroperoxide side product and a THIQ dimer. In the presence of 0.4 mol% [Ru(bpz)3]2+ or 1.0 mol% [Cr(dqp)2]3+, 87% of the 415 nm excitation light was absorbed by the photocatalyst (Supplementary Fig. 109) and consequently some background reactivity involving the direct excitation of THIQ cannot be excluded. In the aza-Henry reaction, the electron donor is not a sacrificial reagent but acts as the substrate, hence the scheme in Fig. 1a does not fully apply. Nonetheless, the initial photoinduced electron transfer from THIQ to excited [Ru(bpz)3]2+ and [Cr(dqp)2]3+ is quantitative given the relevant rate constants (kq, Fig. 2c) and THIQ concentrations (50 mM; Supplementary Section 8.3). The product formation rates were determined to be 1.97 ± 0.12 mM h−1 for [Ru(bpz)3]2+ and 0.56 ± 0.04 mM h−1 for [Cr(dqp)2]3+ (Fig. 4h), corresponding to a ν(RuII)/ν(CrIII) ratio of 3.5 ± 0.3, whereas a ratio of ФP(RuII)/ФP(CrIII) of 3.6 ± 0.5 for THIQ-MeNO2 formation was obtained (Supplementary Fig. 119). These two ratios approach that of the cage escape quantum yields ФCE(RuII)/ФCE(CrIII) > 5.0 for the [Ru(bpz)3]2+–THIQ and [Cr(dqp)2]3+–THIQ donor–acceptor couples (Fig. 4i,j). The cage escape quantum yield for the latter is at the detection limit, hence only a lower limit of 5.0 can be given for the ratio of ФCE values in this case. The photoreaction with [Ru(bpz)3]2+ yielded appreciable amounts of a THIQ dimer (Supplementary Fig. 110), which was not the case with [Cr(dqp)2]3+ (Supplementary Fig. 111). When taking the formation of this THIQ dimer side product into account, an overall reaction rate ratio ν(RuII)/ν(CrIII) of 6.6 ± 1.7 was obtained (Supplementary Fig. 115).
Overall, the key finding from these three photoredox reactions (Fig. 4) is that the initial product formation rates (ν) and the initial product formation quantum yields (ФP) extracted from the photocatalysis experiments correlate with the cage escape quantum yields (ФCE), determined by transient absorption spectroscopy (Fig. 4j).
留言 (0)