
記住我

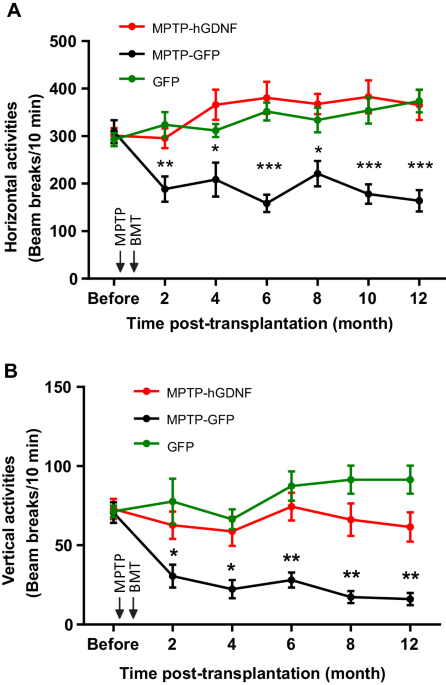
A total of 23 ACT with genetically modified peripheral blood T-cells were identified in the 36 applications received for an OD (Table 1). Of these, 19 (83%) were CAR-T cell products and 4 (17%) were TCR-modified T-cell products. Overall, 19 of the 23 genetically engineered ACT (83%) were granted at least one designation covering 30 different OD. Four products were included in more than one OD application for the treatment of different orphan conditions and have been granted MA for some of them. These were all cluster of differentiation 19 (CD19)-directed autologous CAR-T cell products using either lentiviral- or retroviral vectors developed to treat patients with rare B-cell leukaemia and/or lymphoma. The sponsors of these four CAR-T cell products applied for either up to two, three, four, or eight different OD for each product (Table 1). For two of these products, one OD application each were withdrawn during the regulatory evaluation before an opinion was adopted. An additional two autologous CAR-T cell products using a lentiviral vector and targeting the B-cell maturation antigen (BCMA) were designated for the treatment of multiple myeloma and have been granted a conditional MA.
Table 1 Overview of applications for OD and maintenance of OD at the time of MA evaluated by the COMP for genetically engineered ACT in the period from 2014 to 2023.In total, six (17%) of the OD applications for six products were withdrawn due to insufficient data to fulfil the criteria for an OD. The reasons for the withdrawals were related to (1) failure to provide reliable prevalence estimates based on relevant epidemiologic data sources that showed that the condition (i.e., CLL/SLL) did not breach the prevalence threshold required for an orphan status (1 application), (2) the proposed orphan condition (i.e., HGBCL) was not considered a distinct medical entity, but rather a subtype of a broader condition for which the product already had an OD (1 application), and (3) failure to provide sufficient data to justify the assumption of SB over existing therapies for the proposed conditions (4 applications). It was also noted that the sponsors for three (16%) of the 19 products that were granted either one or several OD withdrew the orphan status for some of the designated conditions, which constituted seven OD in total.
Eighteen (78%) of the products evaluated for OD used a lentiviral vector (16 CAR-T cell products, 2 TCR-modified T-cell products). Only three products used a retrovirus vector (2 CAR-T cell products, 1 TCR-modified T-cell product), one CAR-T cell product used an adeno-associated vector, and one TCR-modified T cell product used messenger RNA. A variety of target antigens were selected for these products with the most common being CD19, which is widely expressed on malignant B-cells, representing 7 (30%) of the 23 products in total, including one bispecific CAR-T cell product targeting both CD19 and CD20. Another common target for 4 (17%) of the products was BCMA, which is expressed on the cell surface of myeloma cells, normal plasma cells, and a small subset of normal B-cells. Other antigens targeted by the genetically engineered ACT included: (2) human leucocyte antigen (HLA)-A2, (2) hepatitis B virus (HBV), (1) signalling lymphocytic activation molecule F7 (SLAMF7), (1) CD1a, (1) CD7, (1) CLDN18.2, (1) CD123 (IL-3R), (1) C-type lectin-like molecule-1 (CLL-1) (CD371), (1) melanoma-associated antigen family A4 (MAGE-A4), and (1) New York oesophageal antigen-1 (NY-ESO-1). The largest group of orphan conditions applied for were haematological conditions, and rare haematological malignancies were designated in 26 (87%) of the 30 OD that were granted, and only 4 OD were given to 4 different products intended to treat either soft tissue sarcoma (2 OD), gastric cancer (1 OD), or solid organ transplantation (1 OD).
There are currently 6 CAR-T cell products approved on the EU market. All six products received both OD and Priority Medicine (PRIME) designations from the COMP/ EMA before the applications for MA were submitted. There were 6 initial MA approved for 10 therapeutic indications, covering 10 different OD, and 4 extensions of the approved indications, covering 3 additional OD (as 1 indication extension was for an earlier treatment line). Three of the authorised CAR-T cell products, i.e., Yescarta, Kymriah, and Breyanzi, received a full approval at the initial MA stage, whereas the other three, i.e., Tecartus, Abecma, and Carvykti, initially received a conditional MA (Table 1A). Conditional MA is recommended by the CHMP when the medicine addresses an unmet medical need and a positive benefit-risk balance for the therapeutic indication is demonstrated, but more comprehensive clinical data to support it in the future is required [20]. All designated products for which maintenance of the OD at the stage of MA or MA extension was applied were developed to treat rare haematological malignancies. None of the two TCR-modified T-cell therapies that were designated for the treatment of soft tissue sarcoma have reached the stage of MA and hence the review of the OD criteria for maintenance of the status.
The most common prevalence of the orphan conditions targeted for in the 36 OD applications received for engineered ACT was estimated to ≥ 3 in 10,000 people in the EU (15/36; 42%), which included multiple myeloma, diffuse large B-cell lymphoma, and follicular lymphoma. The smallest group (9/36; 25%) constituted the orphan conditions with a prevalence of less than 1 in 10,000 people. In total, 12 OD applications (33%) were received for genetically engineered ACT targeting orphan conditions with an estimated prevalence of 1 to < 3 in 10,000 persons. In line with the OD applications, the most common prevalence of the orphan conditions targeted for in the orphan maintenance procedure was ≥ 3 in 10,000 persons (9/14; 64%). For 57% (8/14) of the submissions for maintenance of OD, the prevalence of the targeted orphan conditions had increased since their OD were granted, and the majority of these (6/8) were conditions estimated to be affecting ≥ 3 in 10,000 people in the community.
Large pharmaceutical companies represented the highest number of applicants for OD (22/36; 61%) with small and medium-sized enterprises (SME) and consultancies next (6/36; 17% each). Two OD applications (5%) submitted by each of these three groups of applicants were withdrawn during the regulatory assessment before an opinion was adopted. Only 2 applications for OD (5%) were submitted by academia/charity, and both were granted an OD. In total, 6/30 (20%) granted OD were transferred to another sponsor later. All sponsors that applied for orphan maintenance at the stage of MA or MA extension were large pharmaceutical companies.
Data used to support MP at the initial OD stageOverall, preliminary clinical data was used to support an assumption of MP for the proposed product in 29/36 (81%) OD submissions received for genetically engineered ACT over a 10-years period (Fig. 1). In 25/36 (69%) cases a decision was based purely on clinical data and in 4/36 (11%) on clinical and supporting in vivo data, while in 6/36 (17%) cases the decisions were based only on non-clinical in vivo data. For half of the 6 OD applications where non-clinical in vivo data was the basis for supporting MP, clinical data was also submitted, but considered insufficient for assessment due to limited number of patients. Finally, in 1 submission for OD only in vitro data was used to support MP of the proposed product (Fig. 1).
Fig. 1: Types of data submitted (left) versus those accepted (right) to support the MP criterion in OD applications received for genetically engineered ACT (N = 36).
The number of OD applications that included specific types of data and the proportion of these are given in each coloured box. Data derived from clinical studies is marked in blue, data from both clinical- and non-clinical in vivo studies is marked in orange, data based on non-clinical in vivo studies alone is marked in grey, and pure in vitro data is marked in yellow.
The preliminary clinical data used to support the MP criterion for OD was mainly obtained from single-arm phase 1 or phase 1/2 studies (23/29; 79%) (Fig. 2A). Most of the clinical data in the submissions were collected from small patient populations of < 25 patients (23/32; 72%), but some included data based on observations in populations of ≥ 50 patients (4 applications) (Fig. 2B). Notably, 3 OD applications with clinical data from only one or two patients in compassionate use programmes, and a few additional patients from an investigator-initiated study for one of them, failed to show MP in the proposed orphan conditions. The most common clinical endpoints used to support a potential efficacy of the products were objective anti-tumour responses in terms of overall- (ORR) and complete response rates (CRR) (25/29; 86%) (Fig. 2C).
Fig. 2: Clinical data used to support an assumption of MP for OD applications.
A Overview of cases where clinical data were submitted and those accepted as evidence to support MP and the studies the data were collected from (N = 36). B Summary of numbers of patients, divided into four main categories, which were evaluated in the clinical data provided in the submissions and those accepted as evidence for efficacy to support MP (n = 32). C Overview of the clinical endpoints accepted to support MP in the submissions excluding those that were not supported by clinical data (n = 29). CRR complete response rate, CUP compassionate use programme, DCR disease-control rate (including ORR and stable disease), EFS event-free survival, IIS Investigator-initiated study, None No clinical data was provided in the submissions, ORR objective/overall response rate, PFS progression-free survival.
The non-clinical in vivo data used to show potential efficacy of the candidate products to support MP were derived from valid mouse models of the proposed orphan conditions where the tumour burden and survival of treated mice were evaluated. In all OD applications where these data were accepted (n = 10; 28%), the in vivo data showed reduced tumour burden and prolonged survival of the mice treated with the candidate- or murine surrogate products over control treatments. In vivo tumour xenograft mouse models using non-obese diabetic severe combined immune deficiency IL-2RG (null) (NOD-SCID-gamma null; NSG) mice engrafted with tumours cells from humans with the proposed orphan conditions were the most common models used in the applications where non-clinical data was accepted to support MP. These models were also used in all submissions where only non-clinical in vivo data were used to support the MP criterion (n = 6; 17%).
Data used to support SB at the initial OD stageAll 36 OD applications received for genetically engineered ACT in the period from 2014 to 2023 needed to show that the product applied for could be of SB to those affected by the orphan condition since other satisfactory methods for the proposed condition existed (Fig. 3A). It was noted that the arguments for SB were based on a CRA in 34/36 cases (94%), whereas in two of the submissions the sponsor claimed SB over existing therapies based on a CRA combined with major contribution to patient care. The same data used to support a potential efficacy of the proposed products were used to support an assumption of SB over existing therapies (Fig. 2). Of the 36 submissions for OD, 17 cases (47%) provided data that showed a clinically meaningful benefit in heavily pre-treated patients affected by the orphan condition, 14 (39%) submitted data which indicated improved efficacy over other satisfactory methods for the proposed condition, and 1 (3%) presented data supporting an assumption of both improved efficacy and better safety of the product. The claim of major contribution to patient care was not accepted in any of the two submissions where this was argued by the sponsor, but for one of the applications the claim of SB was accepted based on a CRA in terms of improved efficacy. In total, the sponsors of 4 (11%) of the 36 OD applications submitted failed to provide sufficient data to fulfil the SB criterion. The reasons for these failures were related to insufficient data to support an assumption of SB (1) over other authorised ATMP/innovative therapies demonstrated to be efficacious for the treatment of the proposed condition (i.e., DLBCL; 1 application), (2) limited clinical data (2 applications), or (3) availability of only in vitro data with the proposed product (1 application).
Fig. 3: Arguments for SB (left) versus the claims accepted (right) to support the SB criterion in applications received for OD and maintenance of the status for genetically engineered ACT.
The numbers of submissions were grouped in different coloured boxes according to the arguments given by the sponsors for SB and the claims which were accepted for fulfilling the SB criterion A at the initial OD stage (N = 36) and B at time of review of the criteria for maintenance of OD (N = 14). CRA clinically relevant advantage, E efficacy, MCPC major contribution to patient care, No SB criterion was not considered fulfilled, pts patients, S safety.
Demonstration of SB at time of review of the criteria for maintenance of ODAt the stage of MA and review of the OD criteria, all 14 submissions received for the maintenance of the OD needed to provide data to demonstrate SB as there were at least one and up to six other satisfactory methods approved for the proposed therapeutic indications. Notably, for nine (64%) of these cases, the treatment landscape for the designated orphan condition had changed as one or more other medicines had been approved for the condition since the OD had been granted. In line with the OD applications, the arguments for SB of these products were primarily based on a claim of a CRA in terms of improved efficacy over existing therapies (Fig. 3B). This claim was accepted in most of the cases (12/14; 86%), but in two applications SB was not considered established based on the data presented.
The clinical data used to confirm SB in the applications for maintenance of the OD were derived from the sponsors’ own clinical studies with their CAR-T cell products and mainly from single-arm phase 1 (n = 3), phase 1/2 (n = 2), or phase 2 (n = 8) studies. Only one of the applications which represented an indication extension to an earlier treatment line presented comparative data with the orphan medicinal product from a randomised controled phase 3 study. Most of the data with the proposed products were collected from relatively small patient populations and a few were based on observations from only a limited number of patients with the designated orphan condition (Fig. 4A). The arguments for SB were mainly based on results from a primary endpoint of either ORR or CRR and represented 93% (13/14) of the applications for maintenance of OD (Fig. 4B). The duration of these responses to the new products was considered a key secondary endpoint for demonstrating SB in all these 13 cases. The data used to support SB from the phase 3 study was based on the time-dependent primary endpoint of event-free survival, which was supported by the key secondary endpoint of overall survival.
Fig. 4: Clinical data used to demonstrate SB in applications for maintenance of the OD at time of MA or MA extension (N = 14).
A Overview of numbers of patients and patient populations evaluated in the clinical data derived from the sponsors’ own studies of the new products which were used to justify SB. B The primary- (x-axis) and secondary endpoints (y-axis) of the clinical studies conducted by the sponsors used to support SB. C Numbers of submissions highlighting the type of comparative data presented and those accepted for demonstrating SB. “No” denotes those two cases where SB was not considered fulfilled. ALL acute lymphoblastic leukaemia, CRA clinically relevant advantage, CRR complete response rate, DLBCL diffuse large B-cell lymphoma, EFS event-free survival, FL follicular lymphoma, INV investigator, IRC independent review committee, ITT intent-to-treat (patients enroled), MCL mantle cell lymphoma, mITT modified ITT (patients infused/treated), MM multiple myeloma, ORR objective/overall response rate, OS overall survival, PFS progression-free survival, PMBCL Primary mediastinal large B-cell lymphoma, pts patients, 2 L second-line therapy, 2 L+ second- and later lines of therapy, 3 L+ third- and later lines of therapy, 4 L+ fourth- and later lines of therapy.
The study results from the pivotal single-arm studies were contextualised with published clinical study data as external controls for the satisfactory methods of treatment for the target patient populations to substantiate the SB claim (Fig. 4C). Most submissions included naïve cross-study comparisons, with four also including matching-adjusted indirect comparisons (MAIC) to support a claim for SB of their product. Real-world data from retrospective studies and systematic literature reviews were also submitted in nine of the cases to further contextualise the clinical outcomes reported from the pivotal studies of the new products. In only four of these cases were real-world evidence regarded as sufficient to establish some of the claims for SB, whereas in other four cases they were only used as supporting evidence. One submission which included data from the phase 3 study used the active controlled comparison. Twelve of the 14 submissions were concluded to have provided sufficient data to demonstrate SB, whereas two failed to show a CRA over another approved CAR-T cell product.
Three of 14 submissions were withdrawn before an opinion was adopted and were based on data from one single-arm phase 1 study with the same CAR-T cell product, which was conducted in patients with three designated orphan conditions. It was noted that this product offered a SB for one of the designated orphan conditions, but that the sponsor failed to provide sufficient data to support the claim of SB for the other two conditions. The sponsor decided to withdraw all three OD to stay within the same global MA.
留言 (0)