
記住我

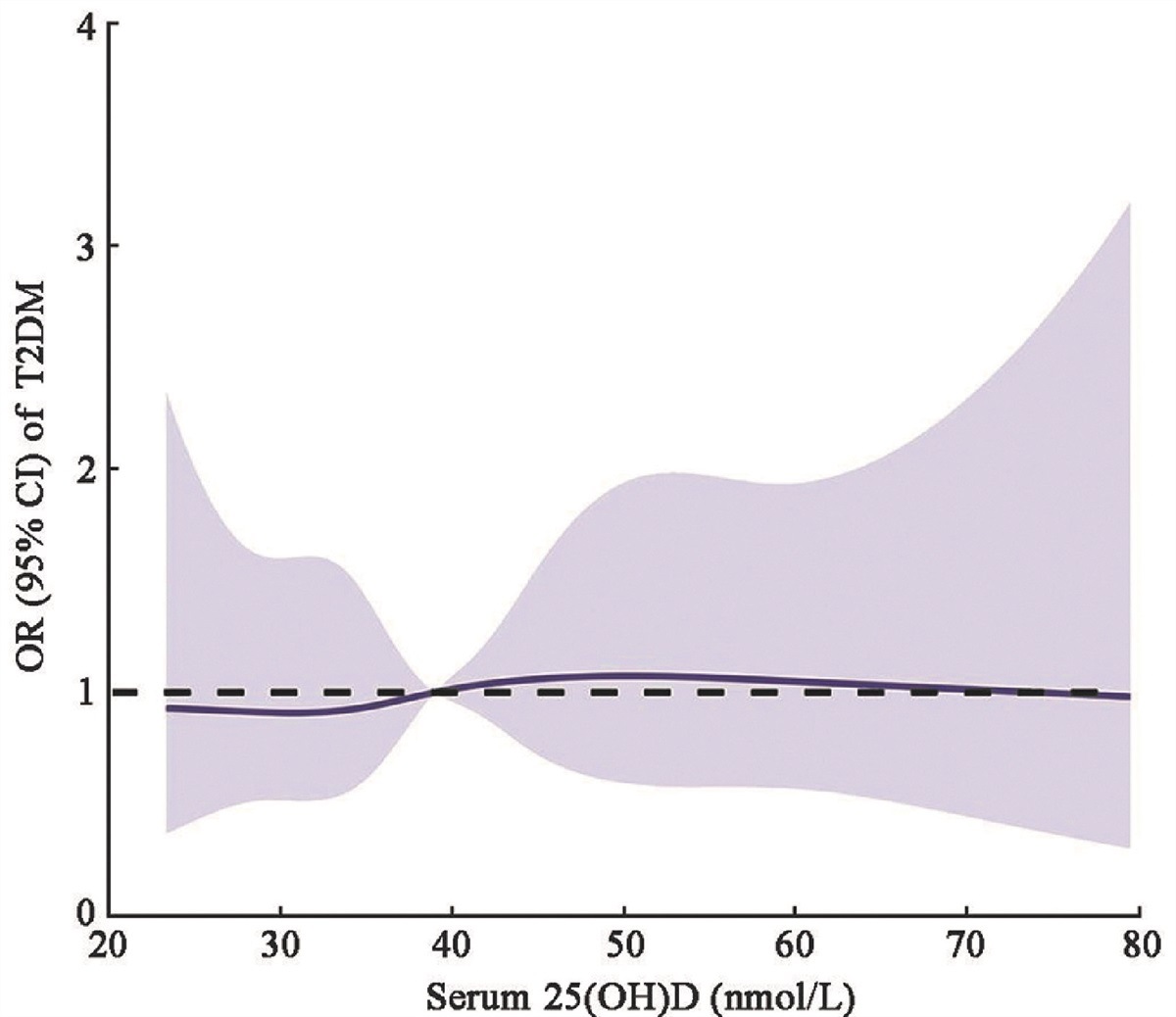





Diabetes mellitus (DM) is a major metabolic disorder that is characterized by a decreased response to insulin and an increase in blood glucose levels. Epidemiologic studies have revealed a strong association between DM and microcirculation dysfunctions (e.g., diabetic retinopathy, diabetic nephropathy, and diabetic myocardiopathy).[1]
Several studies have indicated that DM-induced microvascular disorder is frequently accompanied by endothelial cell apoptosis and abnormal accumulation of the extracellular matrix (ECM) in the subendothelial basement membrane (BM).[2,3] The reduction in endothelial cell number, accompanied by inappropriate elevation of the ECM in the BM, ultimately contributes to insufficient nutrient exchange and further worsens the prognosis of DM patients.[4,5] The ECM is a non-cellular dynamic structure that is made up of various macromolecules, such as collagens, proteoglycans, elastin, fibronectin, and other glycoproteins. The primary function of the ECM is to interact with cells and provide physical support for tissues. In addition, increasing evidence has demonstrated that the ECM also plays an important role in regulating cell viability, growth, differentiation, and metabolism.[6] The components of the ECM are frequently remodeled by surrounding cells via degradation, synthesis, reassembly, and modification.[7] Fibronectin is a multidomain glycoprotein composed of types I, II, and III repeating units. Quiescent blood vessels express less fibronectin, whereas pathological conditions (e.g., diabetes, trauma, atherosclerosis, and myocardial infarction) significantly upregulate the expression of fibronectin.[8,9] Prior studies have reported that fibronectin closely regulates the self-assembly of other ECM proteins and plays an important role in tissue remodeling during the process of DM-induced organ fibrosis.[10,11]
Integrins, which make up a cluster of transmembrane receptors consisting of an α subunit and a β subunit, were proven to be the major receptors of ECM proteins.[12] Pathological studies have demonstrated that the microvasculature of DM patients expresses higher levels of integrin receptors than that of control patients.[13] The increase in integrin and ECM protein levels suggests a potential role of the integrin-ECM interaction in DM-induced microvascular dysfunction.
Previous studies of fibronectin-related integrin ligands have mainly focused on α5β1 and αvβ3, but little is known about the bioeffects of other fibronectin-related integrins. Integrin αvβ5, which is abundantly expressed in human endothelial cells[14] has been demonstrated to interact with fibronectin in prior studies.[15,16] Through biopsies of diabetic human kidneys, it was found that the expression of αvβ5 in glomeruli was elevated in correspondence with the progression of DM, which suggested that αvβ5 may play an important role in DM-induced microvascular injury.[17]
This research sought to investigate the effect of high glucose on the expression of αvβ5 and fibronectin in human vascular endothelial cells. We studied the roles played by fibronectin and αvβ5 in high glucose-induced endothelial cell injury and the underlying mechanisms.
Methods AnimalsThe animal studies were performed in compliance with The Guide for the Care and Use of Laboratory Animals, 8th edition (2011). All the experimental procedures were approved by the Experimental Animals Ethics Committee of Fuwai Hospital (No. FW-2021-0004). Male leptin receptor-knockout (db/db) mice (6 weeks old, C57Bl/6J) and non-diabetic db/m mice were obtained from Cyagen Biosciences (Suzhou, China) and fed normal chow. To selectively delete integrin subunit β5 (ITGB5) from vascular endothelial cells, vascular targeted adeno-associated virus (AAV-Vec, Hanbio Biotechnology Co., Ltd, Shanghai, China) with a specific endothelial cell promoter (TEK receptor tyrosine kinase, TIE2) expressing ITGB5 short hairpin ribonucleic acid (shRNA, miR-30 based) was administered to the mice via tail vein injection (1010 PFU/mice) after one week of adaptation. After 4 weeks of injection, all the mice were sacrificed under deep anesthesia (induced by intraperitoneal injection of sodium pentobarbital at 50 mg/kg), and their plasma and heart were collected for further analysis.
Isolation of endothelial cells from mouse myocardial tissue was performed by using anti-mouse-CD31 microbeads (Miltenyi Biotec GmbH, San Diego, California, USA) as previously described.[18] Briefly, mouse myocardial tissue was washed with cold phosphate buffer solution (PBS) for 3 times and enzymatically dissociated in endothelial cell culture medium (ECCM, Sciencell, Cat. No. 1001, San Diego, California, USA) that contained collagenase/dispase/0.1% trypsin/1.0 mmol/L ethylene diamine tetraacetic acid for 30 min at 37°C. The cells were centrifuged for 5 min at 400 × g and 4°C. The cell pellets were resuspended in ECCM, passed through a Falcon cell strainer (BD, Lake Franklin, New Jeresy, USA) with a pore size of 70 µm, and collected using a 50 mL centrifuge tube. Thereafter, the cells were resuspended in ECCM and sorted with anti-CD31 magnetic beads. CD31+ cells were considered endothelial cells.
Cell culture and treatmentThis study complied with the ethical guidelines of the 1975 Declaration of Helsinki, and was approved by the Ethics Committee of Fuwai Hospital (No. IRB2012-BG-006). All the participants were informed and signed consent form at the time of enrollment. The isolation and identification of human umbilical vascular endothelial cells (HUVECs) were performed in a manner that was consistent with the previously described instructions.[19] HUVECs were maintained in ECCM (manufactured by Sciencell, Cat. No. 1001, San Diego, California, USA) supplemented with 5% fetal bovine serum (FBS), 1% endothelial cell growth supplement (ECGS, manufactured by Sciencell, Cat. No. 1052), and 1% penicillin/streptomycin solution at 37°C in a humid atmosphere with 5% CO2. HUVECs were used in the experimental analyses within 4 passages. High-glucose ECCM and normal-glucose ECCM were made by adding D–(+)-glucose (Sigma Aldrich, G7021, Wisconsin, USA) and glucose-free ECCM (Sciencell), respectively.
Small interfering RNA (siRNA) purchased from GenePharma (Suzhou, China) was used for the transient knockdown of target genes. The nucleotide sequences of siRNAs are listed in Supplementary Table 1, https://links.lww.com/CM9/B622. Lipofectamine RNAiMAX transfection reagent (Invitrogen, California, Carlsbad, USA) was used, and the transfection experiment was performed consistent with the instructions provided by the manufacturer. HUVECs were transfected with siRNAs in Opti-MEMTM (Gibco, Carlsbad, California, USA) for 6 h. After transfection, the medium was replaced by normal culture medium (ECCM supplemented with 5% FBS and 1% ECGS), and the cells were incubated for another 12 h. The interference efficiency was determined by Western blotting analysis.
Cell apoptosis assayAn in situ cell death detection kit (terminal deoxynucleotidyl transferase mediated dUTP nick-end labeling [TUNEL] assay kit, Roche Applied Science, 11767291910, Basel, Switzerland) was used to assess cell apoptosis in vivo and in vitro. Apoptotic cells in the sections of mouse myocardial tissues were stained according to the manufacturer's instructions, and slides were counterstained for endothelial cells using an antibody against CD31. HUVECs were grown in confocal dishes (NEST, Wuxi, China) before TUNEL staining. The experiment was carried out according to the manufacturer's instructions. 4′,6-diamidino-2-phenylindole (DAPI, Abcam, ab104139, Cambridge, UK) was used to stain the cell nuclei. Images were captured by LEICA TCS SP5 MP (Leica, Weztlar, Germany).
Protein extraction and Western blottingAfter being isolated from mouse myocardial tissues, endothelial cells (CD31+) were added to lysis buffer (radio-immunoprecipitation assay [RIPA] buffer containing 1 mmol/L phenylmethanesulfonyl fluoride [PMSF]) to extract the total proteins. The protein concentration of each sample was quantified and equilibrated before Western blotting.
Cells were rinsed twice with cold PBS and collected with a scraper. RIPA buffer (Thermo Fisher, 89901, Waltham Massachusetts, USA) containing 1 mmol/L PMSF (Solarbio, P0100, Beijing, China) was used to extract the total proteins from cells. A protein phosphatase inhibitor (Solarbio, P1260) was added to RIPA buffer when extracting phosphorylated proteins. A nuclear protein extraction kit (Beyotime, P0027, Shanghai, China) was used to extract nuclear proteins from cells, and the experiment was performed according to the manufacturer's instructions. The protein concentration of each sample was equilibrated before the immunoblotting experiments. Antibodies were purchased from Abcam and Cell Signaling Technology (Boston, Massachusetts, USA), and their category number is shown in Supplementary Table 2 [https://links.lww.com/CM9/B622]. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used to normalize the expression of the target protein in each sample. Secondary antibodies were purchased from LI-COR Biosicence (California, San Diego, USA).
Immunofluorescence assay and colocalization analysisMouse myocardial samples were collected and fixed for immunofluorescence analysis. To assess the capillary density in the myocardium, the samples were sliced into 8-μm frozen sections for immunofluorescence staining of CD31.
Cells were grown in confocal dishes (NEST). After rinsing with cold PBS, the cells were fixed with 4% paraformaldehyde for 15 min and incubated with PBS containing 0.3% Triton (Solarbio) for 15 min to permeabilize the membrane. Subsequently, the cells were rinsed twice and incubated with primary antibody overnight. On the next day, the cells were rinsed and incubated with secondary antibody for 45 min. Mounting medium with DAPI (Abcam) was added and incubated with cells for 5 min before analysis. Images were captured by a LEICA TCS SP5 MP, and fluorescence intensity was quantified by ImageJ software (Bethesda, Maryland, USA). The antibodies used for analysis are listed in Supplementary Table 2 [https://links.lww.com/CM9/B622].
We used two different methods to assess the spatial colocalization of two proteins. First, we set the proper area (area of interest, AOI) in pictures where both proteins were well labeled. We used Image-Pro Plus 6.0 (Media Cybernetics Inc, California, USA) to evaluate the Pearson correlation values and overlap coefficient of two proteins in AOI. If Pearson correlation values and overlap coefficients ranged from 0.5 to 1.0, the two proteins were considered to be significantly co-localized.[20] Additionally, we also used one-dimensional analysis to assess the colocalization of two proteins, which has been proven to be a practical method in previous studies.[21]
RNA extraction and real-time quantitative polymerase chain reaction (RT-qPCR) analysisCells were rinsed, and total RNA was extracted by using TRIzol Reagent (Invitrogen). TransScript One-Step gDNA Removal and cDNA Synthesis SuperMix (Transgen Biotech, Beijing, China) was used to conduct reverse transcription, and real-time PCR was performed by applying Top Green qPCR Supermix (Transgen Biotech). Experiments were performed according to the manufacturer's instructions. We used GAPDH to standardize the expression of target genes, and the primer sequences are shown in Supplementary Table 3, https://links.lww.com/CM9/B622. We quantified the expression of target genes by calculating 2-ΔΔCt values.
Lentiviral transfection processAfter isolation from tissues, HUVECs were incubated in normal medium for 48 h before transfection with lentivirus. To detect the autophagic flux in HUVECs, we used green fluorescent protein(GFP)-monomeric red fluorescent protein (mRFP)-microtubule associated protein light chain 3 (LC3) lentivirus (Hanbio Biotechnology Co, Ltd) to label autophagosomes and monitor their formation and degradation. To establish a stable FoxO1-overexpressing HUVEC cell line, we used flag-PURO-FoxO1 lentivirus (Hanbio Biotechnology Co, Ltd) to infect HUVECs at an multiplicity of infection (MOI) of 30 (with 4 µg/mL polybrene) and selected stable transfectants by adding puromycin at a concentration of 2 µg/mL. Immunoblotting of the Flag tag was used to validate the successful transfection of FoxO1.
RNA sequencing (RNA-seq) analysisRNA-seq and analysis were performed by Annoroad Gene Technology Co. (Beijing, China), and the Illumina platform (San Diego, California, USA) was used. The concentration and quality of all the samples were determined before the analysis. Individual cDNA libraries were constructed by using RNA samples. Cluster generation and sequencing were performed on the NovaSeq 6000 S4 platform using the NovaSeq 6000 S4 Reagent kit V1.5 (Illumina, San Diego). The false discovery rate (FDR) was calculated, and deferential expression of genes was determined by using DESeq2 algorithms.[22] The P-value of each gene was calculated and adjusted by Benjamini and Hochberg's approach for controlling the FDR (q value). Genes with q ≤0.05 and |fold change| ≥1 were defined as differentially expressed genes (DEGs). The rich ratio of pathways was quantified by using Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis. The raw RNA-seq data were uploaded to the Sequence Read Archive (SRA) database under accession number PRJNA931645.
Statistical analysisThe results are presented as the mean ± standard deviation (SD) for continuous data. We performed the Kolmogorov–Smirnov test to determine whether the data conformed to a normal distribution. The Kruskal–Wallis test was used to determine if means across groups were significantly different before reporting analysis of variance (ANOVA)-associated post hoc test results. One-way ANOVA followed by Bonferroni multiple comparisons post hoc test was performed to assess the significance of the deviation when three or more groups were involved. For data that did not follow a normal distribution, non-parametric tests (Wilcoxon rank test) were used. The statistical methods used in graphs are described in the Supplementary Table 4 [https://links.lww.com/CM9/B622]. All the data were analyzed by GraphPad Prism 7 (San Diego, California, USA). P-values <0.05 were considered statistically significant. Representative images were selected as those that showed values close to the means of the results obtained from all analyzed samples.
Results Hyperglycemia promoted endothelial apoptosis and reduced capillary density in the myocardium of diabetic mice, whereas knockdown of ITGB5 attenuated these effectsCompared with db/m mice, fasting blood glucose levels significantly increased in db/db mice. Selective knockdown of ITGB5 in vascular endothelial cells did not have a significant effect on fasting blood levels in db/db mice [Table 1]. Immunoblotting analysis of vascular endothelial cells isolated from the myocardium revealed that the expression of fibronectin, integrin αv (ITGAV), and ITGB5 was increased in the vascular endothelium of diabetic mice [Figures 1A–D]. Protein expression analysis also indicated that the expression of ITGB5 was successfully knocked down by using an AAV-Vec-based ITGB5 shRNA delivery strategy [Figures 1A,D]. In addition, macroautophagy (hereafter referred to as autophagy) activity in endothelial cells was observed to be elevated under hyperglycemic conditions, whereas knockdown of ITGB5 significantly attenuated autophagy [Figures 1A,E]. These observations suggested that integrin may play a role in regulating hyperglycemia-associated endothelial injury.
Table 1 - Characteristics of db/m mice, db/db mice and db/db mice with ITGB5 shRNA injection (db/db+ITGB5 shRNA). Characteristics db/m db/db db/db + ITGB5 shRNA n 11 11 11 Male (%) 100 100 100 Age (weeks) 12 12 12 Body weight (g) 24.1 ± 0.7 37.6 ± 0.6* 36.6 ± 0.6* Fasting blood glucose (mg/dL) 173.6 ± 5.0 454.1 ± 16.3* 448.5 ± 15.1**P <0.0001 vs. db/m. Data are expressed as n or mean ± standard deviation. Baseline characteristics of experimental animals were recorded. The 8-week gene-modified mice was subjected to AAV-based ITGB5 shRNA delivery through tail vein injection. Four weeks after injection, their blood samples were collected and analyzed. AAV: Adeno-associated virus; ITGB5: Integrin subunit β5; shRNA: Short hairpin ribonucleic acid.
 Figure 1:
Figure 1: Assessment of diabetic mice. Some of eight-week gene-modified mice were subjected to AAV-based ITGB5 shRNA delivery through tail vein injection. Four weeks after injection, myocardial samples were collected and analyzed. (A) Mouse cardiac vascular endothelial cells were isolated from the myocardium and subjected to Western blotting analysis. (B–E) Relative gray values were used to indicate the expression intensity of target proteins (n = 4/group). (F) Mouse myocardial samples were stained with immunofluorescent antibodies to determine the expression of the target protein (n = 7/group). CD31 (the marker of endothelial cells) was stained red. Apoptotic cells were detected by TUNEL assay (green). The nuclei of the cells were stained with DAPI (blue). The apoptotic endothelial cells were visualized by overlapping the red signal, green signal, and blue signal (marked by arrows). (G) The capillary density of the myocardium was determined by calculating the number of capillaries per square millimeter (NA/mm2). (H) The degree of endothelial cell apoptosis is expressed as the percentage of TUNEL-positive endothelial cells. In the graphs, the data are expressed as the mean ± standard deviation. *P <0.05, †P <0.001, ‡P <0.01. AAV: Adeno-associated virus; DAPI: 4′, 6-diamidino-2-phenylindole; ECs: Endothelial cells; FBN: Fibronectin; ITGAV: Integrin subunit αv; ITGB5: Integrin subunit β5; LC3: Microtubule associated protein light chain 3; n.s: Not significant; shRNA: Short hairpin ribonucleic acid; TUNEL: Terminal deoxynucleotidyl transferase mediated dUTP nick-end labeling.
We further investigated the effects of hyperglycemia on capillary density and endothelial cell apoptosis in the myocardium of diabetic mice. Endothelial cells were labeled with a CD31 antibody and dyed red [Figure 1F]. Immunofluorescence analysis of myocardial tissue indicated that capillary density was markedly decreased in diabetic mice; however, knocking down ITGB5 expression in endothelial cells partially rescued hyperglycemia-induced capillary rarefaction [Figures 1F,G]. The nuclei of apoptotic cells were dyed green using a TUNEL assay kit [Figure 1F]. Apoptotic endothelial cells were detected by overlapping the green signal and red signal [Figure 1F]. The results revealed that hyperglycemia promoted endothelial cell apoptosis, whereas knockdown of ITGB5 alleviated apoptosis in endothelial cells [Figures 1F,H]. By using an animal model of diabetes, we demonstrated that ITGB5 played an important role in mediating hyperglycemia-induced endothelial injury.
High glucose induced the overexpression of integrin αvβ5 and fibronectin in HUVECsTo explore the effects of high glucose on endothelial cells, we incubated HUVECs under normal glucose (5 mmol/L) or high glucose (33 mmol/L) conditions for different lengths of time. Total RNA and protein were extracted from cells and subjected to RT-qPCR [Supplementary Figures 1A–C, https://links.lww.com/CM9/B622] and immunoblotting analysis [Supplementary Figure 1D, https://links.lww.com/CM9/B622]. The results indicated that after stimulation with high glucose for 6 h, 24 h, and 48 h, the expression of fibronectin and ITGAV was significantly elevated [Supplementary Figures 1A,B,E,F, https://links.lww.com/CM9/B622]. However, in comparison with the control group, the expression of ITGB5 in the high glucose-treated group only started to increase after 24 h of stimulation [Supplementary Figures 1C, D, G, https://links.lww.com/CM9/B622]. In addition, to investigate the expression level of the αvβ5 heterodimer (consisting of ITGAV and ITGB5) and fibronectin, we performed immunofluorescence microscopy [Supplementary Figure 1H, https://links.lww.com/CM9/B622]. The results we obtained from immunofluorescence analysis corresponded with those from RT-qPCR and immunoblotting, which indicated that expression of fibronectin started to rise after 6 hours of incubation [Supplementary Figure 1I, https://links.lww.com/CM9/B622], while αvβ5 started to increase after 24 h of incubation [Supplementary Figure 1J, https://links.lww.com/CM9/B622].
Previous studies have demonstrated that fibronectin can bind to integrin αvβ5. To determine whether fibronectin colocalized with αvβ5 in HUVECs under the experimental conditions, we performed immunofluorescence analysis. We set the proper area for colocalization analysis and quantified the Pearson correlation values (Rr = 0.720453) and overlap coefficient (R = 0.803709), which suggested that fibronectin significantly colocalized with αvβ5 in HUVECs [Supplementary Figure 1K, https://links.lww.com/CM9/B622]. One-dimensional analysis of the representative area also indicated obvious colocalization of these two proteins [Supplementary Figure 1L, https://links.lww.com/CM9/B622].
High glucose-induced overexpression of fibronectin promoted apoptosis in HUVECs through ITGB5To validate the role played by the fibronectin–αvβ5 interaction in high glucose-induced cell apoptosis, we assessed the expression of apoptosis-related proteins. The results showed that after incubation under high glucose conditions for 24 h, the expression level of cleaved caspase-3, which is an initiator of the apoptotic pathway, was significantly elevated [Figures 2A,B]. Of note, the trend in cleaved caspase-3 expression was similar to that of the αvβ5 heterodimer and ITGB5, which suggested underlying associations between these proteins. After knocking down the expression of fibronectin, the elevation of cleaved caspase-3 induced by high glucose was significantly inhibited [Figures 2A,B]. These results indicated that the fibronectin–αvβ5 axis could play an important role in high glucose-induced endothelial cell apoptosis.
 Figure 2:
Figure 2: High glucose-induced overexpression of fibronectin promoted apoptosis in HUVECs through ITGB5. (A) After transfection with negative control siRNA or fibronectin siRNA, HUVECs were incubated in high-glucose ECCM (containing 0.2% FBS and 33 mmol/L glucose) or normal-glucose ECCM (containing 0.2% FBS and 5 mmol/L glucose) for 6 h, 24 h, and 48 h. Protein was extracted from each sample and subjected to Western blotting analysis (n = 3/group). (B) Relative gray values were used to indicate the expression intensity of target proteins. (C) In control group, HUVECs were transfected with negative siRNA and then incubated in normal-glucose ECCM for 24 h. And other HUVECs were transfected with negative control siRNA, ITGB5 siRNA, or ITGAV siRNA and then incubated in high-glucose ECCM for 24 h. Protein from each sample was extracted and subjected to Western blotting analysis (n = 4/group). (D–F) Relative gray values were used to indicate the expression intensity of target proteins. (G) In control group, HUVECs were transfected with negative siRNA and then incubated in vehicle-containing ECCM for 6 h. And other HUVECs were transfected with negative control siRNA, ITGB5 siRNA, or ITGAV siRNA and then incubated in fibronefctin-containing ECCM (5 µg/mL fibronectin, 0.1% FBS, 5 mmol/L glucose) for 6 h. Protein was extracted from each sample and subjected to Western blotting analysis (n = 4/group). (H–J) Relative gray values were used to indicate the expression intensity of target proteins. (K) In control group, HUVECs were transfected with negative siRNA and then incubated in normal-glucose ECCM for 24 h. And other HUVECs were transfected with negative control siRNA, ITGB5 siRNA, or ITGAV siRNA and then incubated in high-glucose ECCM for 24 h. Samples were subjected to TUNEL staining (n = 6/group, TUNEL-positive nuclei were marked by arrows). (L) The apoptosis rate was quantified by calculating the percentage of TUNEL-positive cells. (M) In control group, HUVECs were transfected with negative siRNA and then incubated in vehicle-containing ECCM for 6 h. And other HUVECs were transfected with negative control siRNA, ITGB5 siRNA, or ITGAV siRNA and then incubated in fibronefctin-containing ECCM (5 µg/mL fibronectin, 0.1% FBS, 5 mmol/L glucose) for 6 h. Samples were subjected to TUNEL staining (n = 6/group, TUNEL-positive nuclei are marked by arrows). (N) The apoptosis rate was quantified by calculating the percentage of TUNEL-positive cells. In the graphs, the data are expressed as the mean ± standard deviation. *P <0.01, †P <0.001. BAX: B-cell lymphoma-2 associated X; BCL-2: B-cell lymphoma-2; DAPI: 4′,6-diamidino-2-phenylindole; ECCM: Endothelial cell culture medium; FBN: Fibronectin; FBS: Fetal bovine serum; GAPDH: Glyceraldehyde 3-phosphate dehydrogenase; HG: High-glucose medium; HUVECs: Human umbilical vascular endothelial cells; ITGAV: Integrin subunit αv; ITGB5: Integrin subunit β5; KD: Knockdown; NC: Negative control siRNA; NG: Normal-glucose medium; n.s.: Not significant; siRNA: Small interfering RNA. TUNEL: Terminal deoxynucleotidyl transferase mediated dUTP nick-end labeling.
To determine which subunit was indispensable in triggering cell apoptosis, we used gene intervention technology to knock down the ITGAV or ITGB5. Immunoblotting analysis revealed that knockdown of the ITGB5 reduced the high glucose-induced or fibronectin-induced elevation of proapoptotic proteins (cleaved caspase-3 and bax) and increased the expression of an anti-apoptotic protein (bcl-2) [Figures 2C–J]. However, knockdown of the ITGAV did not significantly inhibit cell apoptosis under identical conditions [Figures 2C–J]. In addition to protein expression analysis, we also used TUNEL staining to investigate apoptosis at the cellular level. The results were consistent with those from immunoblotting analysis [Figures 2K–N]. Combined with the protein expression and TUNEL staining results, we discovered that expression of the ITGB5 is critical for both high glucose-induced and fibronectin-induced HUVEC apoptosis.
ITGB5 knockdown alleviated autophagy induced by high glucose and fibronectinIt has been reported that autophagy, which is a cellular mechanism for combating stress-induced damage, plays a vital role in high glucose-induced cell apoptosis.[22] Therefore, we further investigated the effects of ITGB5 on cell autophagy activity. Chloroquine (CQ) was used to block autophagosome degradation so that we could monitor the intensity of the autophagic flux. Through immunoblotting analysis, we found that both high glucose and fibronectin stimulated the expression of beclin1 (the initiator of autophagy) and degradation of LC3 II [Supplementary Figure 2, https://links.lww.com/CM9/B622]. However, we also discovered that the expression of Atg7 (which mainly regulates the maturation of autophagosomes) remained unchanged [Supplementary Figure 2, https://links.lww.com/CM9/B622]. The results above suggested that both high glucose and fibronectin can promote the formation of autophagosomes in endothelial cells, but the ability of these cells to process autophagosomes was not elevated accordingly. After knocking down the expression of ITGB5, the level of beclin1 was downregulated, and degradation of LC3 II was restricted, while the expression of Atg7 was unaffected [Supplementary Figure 2, https://links.lww.com/CM9/B622].
In addition, we also constructed an GFP-mRFP-LC3-expressing cell line to monitor the transition of autophagosomes to autolysosomes. The early-stage autophagosome was labeled by both mRFP and GFP. After being fused with lysosomes, the autophagosome changed into an autolysosome, and GFP was degraded, which made it only labeled by mRFP. The fluorescence microscopy results indicated that autophagosome transition was active in both the high glucose-treated and fibronectin-treated groups [Supplementary Figure 2, https://links.lww.com/CM9/B622]. After knockdown of the ITGB5, the autophagic flux was significantly inhibited under both conditions [Supplementary Figure 2, https://links.lww.com/CM9/B622]. The results further indicated that fibronectin-β5 was involved in autophagy regulation under high glucose stimulation.
FoxO1 was a downstream target regulated by integrin β5To identify the downstream signaling pathway regulated by integrin β5, we conducted RNA-seq analysis and further validated the results by immunoblotting experiments. According to our hypothesis, high glucose elevated the expression of fibronectin in HUVECs, which then stimulated cell apoptosis via integrin β5. Therefore, we designed two cell models in which apoptosis was activated by high glucose or fibronectin. After knocking down ITGB5 in both models, we used RNA-seq technology to determine the gene expression profiles of the samples. KEGG pathway analysis was used to identify differentially regulated pathways in the ITGB5-knockdown groups. Sample clustering analysis showed good intragroup consistency [Supplementary Figure 3, https://links.lww.com/CM9/B622]. The number of differentially regulated genes is shown in Supplementary Figure 4 [https://links.lww.com/CM9/B622]. FDR was evaluated and used to assess the significance of differences in pathway enrichment between ITGB5-knockdown groups and control groups. The pathways with FDR less than 0.05 were considered to be significant [Supplementary Figure 5, https://links.lww.com/CM9/B622]. We screened 10 pathways with the most significant differences according to the FDR ranking, and we arranged them through their rich ratio. The advanced glycation end products (AGE)-receptor of advanced glycation end product (RAGE) signaling pathway showed a significant difference after knocking down the expression of the ITGB5 in both models [Supplementary Figure 6, https://links.lww.com/CM9/B622]. Then, we analyzed the expression levels of genes regulated by the AGE-RAGE pathway. The results indicated that after knocking down the expression of ITGB5 in both models, AGE-RAGE pathway activity was attenuated [Supplementary Figure 6, https://links.lww.com/CM9/B622]. Furthermore, we selected 10 genes regulated by the AGE-RAGE pathway and performed RT-qPCR analysis to validate their expression levels. The results also demonstrated that the knockdown of ITGB5 markedly reduced the activity of the AGE-RAGE pathway [Supplementary Figure 6, https://links.lww.com/CM9/B622]. On the basis of the RNA-seq and RT-qPCR results, it was suggested that ITGB5 was a potential regulator of the AGE-RAGE pathway. Previous research indicated that FoxO1 is an important transcription factor that is regulated by the AGE-RAGE pathway, which controls the autophagic flux and cell apoptosis under high-glucose conditions.[23]
To further validate the RNA-seq results, we subsequently designed experiments to examine the effects of ITGB5 on the functions of FoxO1. Immunoblotting analysis indicated that high glucose and fibronectin both induced FoxO1 expression and nuclear translocation [Supplementary Figure 7, https://links.lww.com/CM9/B622]. However, after knocking down ITGB5, these bioeffects were significantly ablated [Supplementary Figure 7, https://links.lww.com/CM9/B622]. Immunofluorescence microscopy showed that FoxO1 nuclear translocation was markedly activated after stimulation with high glucose or fibronectin, and the knockdown of ITGB5 strongly ameliorated these effects [Supplementary Figure 7, https://links.lww.com/CM9/B622].
FoxO1 regulated the excessive autophagy induced by high glucose and fibronectin, which contributed to HUVEC apoptosisWe further investigated the role of autophagy in high glucose-induced and fibronectin-induced HUVEC apoptosis. On the basis of the autophagy-related protein assay that we conducted, beclin1 was correlated with the elevation of autophagy under experimental conditions. In addition, we also sought to validate the association between FoxO1 and cell apoptosis induced by high glucose and fibronectin. The knockdown of either FoxO1 or beclin1 markedly inhibited the expression of pro-apoptotic proteins (cleaved caspase-3 and bax) and elevated anti-apoptotic protein (bcl2) [Supplementary Figure 8, https://links.lww.com/CM9/B622]. The TUNEL analysis also indicated that both FoxO1 knockdown and beclin1 knockdown attenuated cell death under the provided conditions [Supplementary Figure 8, https://links.lww.com/CM9/B622]. The above results indicated that beclin1-related autophagy played a deleterious role in the apoptosis elicited by high glucose or fibronectin. In addition, the knockdown of FoxO1 significantly reversed the beclin1 overexpression and autophagy that were induced by high glucose or fibronectin [Supplementary Figure 9, https://links.lww.com/CM9/B622], which indicated that beclin-1-related autophagy was regulated by FoxO1.
Overexpression of FoxO1 reversed the effects of ITGB5 knockdownTo determine whether FoxO1 was a downstream target by which ITGB5 regulates cell apoptosis, we constructed a FoxO1-overexpressing cell line. The immunoblotting of the Flag tag indicated the successful transfection of FoxO1 [Figure 3]. The detection of cleaved caspase-3 revealed that overexpression of FoxO1 in HUVECs significantly reduced the anti-apoptotic effects of ITGB5 knockdown [Figure 3]. In addition, after knocking down ITGB5, the apoptotic rate in the empty vector-transfected group was reduced [Figure 3], while FoxO1-overexpressing HUVECs did not exhibit a significant difference [Figure 3]. We also investigated the effects of FoxO1 overexpression on ITGB5-mediated autophagy. The immunoblotting results showed that the empty vector did not affect ITGB5 knockdown-induced reduction in autophagy [Figure 4]. However, the overexpression of FoxO1 markedly reversed the decrease in autophagy induced by ITGB5 knockdown [Figure 4]. The graphical abstract of this research was shown in Figure 4G.
 Figure 3:
Figure 3: The overexpression of FoxO1 in HUVECs reduced the protective effects of ITGB5 knockdown under high glucose conditions. FoxO1-overexpressing HUVECs and empty vector-infected HUVECs were transfected with negative control siRNA or ITGB5 siRNA, and both were stimulated with high-glucose medium for 24 h. (A) Western blotting analysis was used to measure the expression of target proteins. (B,C) Relative gray values were used to indicate the expression intensity of target proteins (n = 4/group). (D) TUNEL analysis was used to analyze cell death. The nuclei of dead cells were stained green (n = 6/group, TUNEL-positive nuclei are marked by arrows). (E) The apoptosis rate was quantified by calculating the percentage of TUNEL-positive cells (n = 6/group). (F) TUNEL analysis was used to analyze cell death. The nuclei of dead cells were stained green (n = 6/group, TUNEL-positive nuclei are marked by arrows). (G) The apoptosis rate was quantified by calculating the percentage of TUNEL-positive cells (n = 6/group). In the graphs, the data are expressed as the mean ± standard deviation. *P <0.001. DAPI: 4′, 6-diamidino-2-phenylindole; FoxO1: Forkhead Box Protein O1; HG: High-glucose medium; HUVECs: Human umbilical vascular endothelial cells; ITGB5: Integrin subunit β5; KD: Knockdown; n.s.: Not significant; NC: Negative control siRNA; NG: Normal-glucose medium; siRNA: Small interfering RNA; TUNEL: Terminal deoxynucleotidyl transferase mediated dUTP nick-end labeling.
 Figure 4:
Figure 4: The overexpression of FoxO1 reversed the anti-autophagy effects of ITGB5 knockdown under high glucose conditions. FoxO1-overexpressing HUVECs and empty vector-infected HUVECs were transfected with negative control siRNA or ITGB5 siRNA and then stimulated with high-glucose medium for 26 h. In several groups, CQ (50 µmol/L) was added to the medium in the last 2 h of incubation. (A) Western blotting was used to measure the expression of target proteins in samples. (B,C) Relative gray values were used to indicate the expression intensity of target proteins (n = 4/group). (D) Western blotting was used to measure the expression of target proteins in samples. (E,F) Relative gray values were used to indicate the expression intensity of target proteins (n = 4/group). ∆LC3 II represents LC3 II degradation, which positively correlates with the autophagy flux. (G) The graph shows that the ITGB5–FoxO1 axis regulated excessive autophagy induced by high glucose and reduced autophagy-related cell apoptosis in HUVECs. In the graphs, the data are expressed as the mean ± standard deviation. *P <0.001, †P <0.01. CQ: Chloroquine; FoxO1: Forkhead Box Protein O1; GAPDH: Glyceraldehyde 3-phosphate dehydrogenase; HG: High-glucose medium; HUVECs: Human umbilical vascular endothelial cells; ITGB5: Integrin subunit β5; KD: Knockdown; LC3: Microtubule associated protein light chain 3; NC: Negative control siRNA; NG: Normal-glucose medium; n.s.: Not significant; siRNA: Small interfering RNA.
DiscussionOur research indicated that ITGB5 played an important role in regulating hyperglycemia-induced endothelial cell apoptosis. Animal research revealed that after stimulation with high glucose, endothelial cell apoptosis was elevated and capillary density was significantly reduced in diabetic mice. After knocking down the expression of ITGB5 in endothelial cells, t
留言 (0)