
記住我



Alzheimer’s disease (AD) is the most common neurodegenerative disease in the United States (US). Often found in elderly patients, aged 65 and above, it is characterized by memory loss, confusion, and behavioral changes [1]. It is also the 6th leading cause of death in the US and the leading cause of death in elderly. In 2021, the healthcare costs for AD patients in the US are about $355 billion and expected to be increased to $1.1 trillion in 2050 [2]. Despite the encouraging results from recent clinical trials of anti-amyloid antibody therapies [3, 4], the modest beneficial effects at slowing the rate of cognitive decline with significant side-effects indicate the urgent needs of developing more efficacious disease modifying therapies for AD.
The cardinal features of AD pathology are amyloid plaques due to excessive Aβ accumulation and neurofibrillary tangles (NFT) due to tau hyperphosphorylation [5]. These changes lead to neuronal death and cortical volume loss [1]. Besides autosomal dominant mutations of three genes (APP, PSEN1 and PSEN2) identified in familial AD patients, there are risk factors associated with late-onset AD (LOAD) such as Apolipoprotein E4 (APOE4) and Triggering Receptor Expressed on Myeloid Cells 2 (TREM2). The APOE4 allele is the strongest risk factor associated with AD. Conversely the APOE2 allele is the strongest genetic protective factor against AD [6]. TREM2 is a receptor involved in the function of microglia in the CNS and variants of TREM2 have been associated with an increased risk of developing AD. It is suggested that TREM2 is vital in the microglial role in phagocytosis of cellular debris like Aβ [7]. Animal models, specifically mouse models carrying various AD risk genes such as APOE4 and TREM2 variants, have been developed to better elucidate disease mechanisms and test therapeutic strategies.
Generally, animal models aim to display the key pathological features of AD such as amyloid and tau pathology, as well as synaptic and neuronal degeneration. In addition, AD pathology should be developed in areas of the brain in a predictable way, similarly to disease progression in human AD patients. More importantly, cognitive function deterioration should follow human disease trajectory. A large portion of effort has been focused on developing transgenic (Tg) mouse models through over-expression of genetic mutations associated with familial AD (FAD) patients [8]. While some AD pathology such as amyloid plaques, neuroinflammation and cognitive impairment can be captured in these mouse models, many fail to display significant degeneration and neuronal loss. Newer generations of mouse models through knock-in (KI)/knock-out (KO) or Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) gene editing technologies, have been developed with the hope to more accurately model proteinopathies without over-expression of human AD genes in mouse brains.
In this review, we summarized a few well-established and commonly used mouse models, as well as newly developed mouse models of AD developed in translational research laboratories today, including both the traditional Tg mouse models, the new KI/KO models and other mouse models such as SAMP8 and seeding models (Fig. 1). For Tg mouse models, we included Tg2576, TgCRND8, APP/PS1, 5xFAD, and 3xTg-AD. We also discussed tauopathy models such as P301S, rTg4510 and P301L. For non-transgenic mouse models, APP-KI, Tau-KI, human APOE KI, TREM2-KO, hTREM2 KI and TREM2 Tg mouse models were discussed. The advantages and limitations of some of these AD mouse models have been recently discussed [9]. In this review, we further compared newly developed AD mouse models (e.g. TREM2 KO/KI and Tg mouse models, the MODEL-AD consortium LOAD mouse models, SAMP8 and seeding models) with previously well-established mouse models, including phenotypic characterization along with discussions of any sex-specific features (as summarized in Tables 1 and 2). More importantly, the publicly available transcriptomics data of various AD mouse models have been analyzed to categorize molecular signatures of each mouse model reminiscent of human AD brain changes (Figs. 2 and 3). Our proof-of-concept analyses compare phenotypic characterization with molecular signatures of mouse models in alignment with human AD brain signature changes, with the hope to guide our future effort to better characterize AD molecular and phenotypic signatures in new-generation mouse models and more importantly, to direct our selection of best mouse models for specific research questions to be addressed in the field. Finally, future needs of developing novel model systems for AD have been discussed such as developing new mouse models carrying novel AD risk variants to be identified or novel model systems to capture heterogenous disease mechanisms of AD such as vascular and environmental contributions, as well as human and mouse chimeric system to incorporate human inducible pluripotent stem cell (iPSC) systems into mouse models through transplantation approaches (Fig. 1).
Fig. 1
Summary of past, present and future in vivo model studies of AD. Traditional mouse models used in the past included various transgenic and KI/KO mouse models with phenotypic characterization of neuropathological and behavior changes. With new information gathered about novel risk variants of AD as well as new AD biomarkers, novel tools have been developed to deep phenotype many of existing mouse models of AD including EEG/sleep studies, neuro-imaging modalities as well as multi-omics analyses. The integration of mouse and human datasets facilitate a better understanding of molecular signatures of each mouse model reminiscent of human AD brain molecular signatures. Future directions such as developing new mouse models carrying novel AD risk variants to be identified, as well as human and mouse chimeric system to incorporate human iPSC systems into mouse models through transplantation approaches may provide novel insights into future in vivo modeling of AD
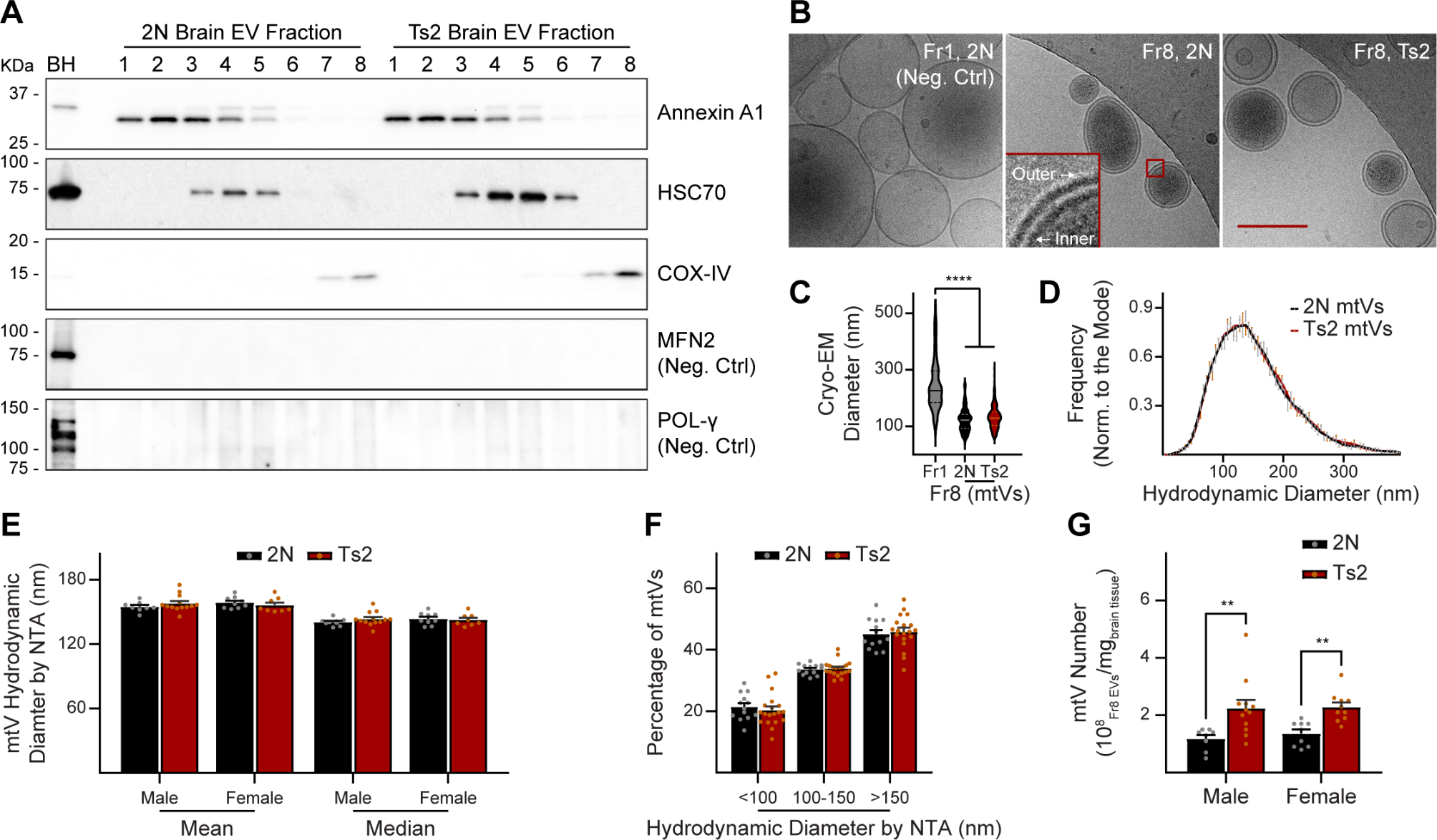
Table 1 Summary of 8 commonly used AD transgenic mouse modelsTable 2 Summary of 6 KI/KO mouse models of ADFig. 2
Comparison of molecular gene signatures between AD mouse model datasets and human AD brain datasets. Heatmap shows the p value significance (-Log10adjusted p values) of the overlaps between AD mouse model molecular signatures and human brain molecular signatures in relate to multiple cognitive/pathological traits (plaque means, CDR, Braak score, CERAD) derived from the PHG brain regions of the Mount Sinai Brain Bank (MSBB) cohort. Axis labels denote the trait contrasts for each gene signature overlap between mouse and human studies. Down- and up-regulated gene signatures are labeled in blue and red colors, respectively
Fig. 3
Comparison of gene ontology (GO)/pathways between the AD mouse model datasets and human AD brain datasets. Sanky network plots show the commonly shared GO/pathways between mouse (left) and human (right) gene signatures. Each node represents a gene signature or a GO/pathway term. Each link colored based of individual GO/pathway term represents a significant overlap between mouse and human gene signatures. A Commonly shared GO/pathways involved in amyloid processes in AD; B Commonly shared GO/pathways involved in neuroinflammation and immune responses in AD
Transgenic mouse models of Alzheimer’s diseaseAmyloid Tg mice Tg2576The Tg2576 model expresses the 695-amino acid isoform of human amyloid precursor protein (APP) with the Swedish mutation inserted into the hamster prion protein (PrP) coplasmid vector resulting in a fivefold increase in Aβ40 and a 14-fold increase in Aβ42/Aβ40. Around 6–7 months of age, Tg2576 mice were found to accumulate Aβ40 and Aβ42 species that were Sodium Dodecyl Sulfate (SDS)-resistant. At 7–8 months of age, amyloid plaques became dense and visible with a wide spread of plaque build-up and deposit on mouse brain parenchyma as well as vascular structures by 11–13 months of age [10]. It was also found that female Tg2756 were more susceptible to developing plaques than their male counterparts [9].
Unlike most other APP models, cognitive decline in Tg2576 mice manifested months prior to pathology whereas cognitive decline occurred in close proximity in other models [8]. Cognitive impairment in Tg2576 mice can be manifested as impaired spatial and working memory measured by behavioral tasks such as the Y-maze, the Morris water maze and the contextual fearing conditioning tests [10,11,12]. Some reported cognitive deficits as early as 6 months of age but most studies reported cognitive dysfunction starting 9–10 months of age and progressively noticeable after 12-month of age [10,11,12]. One study reported a decreased frequency of burrowing in Tg 2576 mice that can be seen as early as 3 months of age preceding the formation of amyloid plaques [13]. Sex-specific differences in cognitive impairment were noted with a rapid progression in females [14], as well as a greater degree of cognitive impairment observed in female mice [9] with the Tg2576 mouse model.
Besides cognitive deficits, other behavior disturbance was reported in the Tg2576 mouse model such as non-rapid eye movement (NREM) sleep disorder and an increased susceptibility to seizure. At 22 months of age, male Tg2576 mice had disrupted sleep EEG rhythms and female Tg2576 mice had decreased REM sleep and delayed sleep onset [15]. It was speculated that cholinergic dysfunction may contribute to sleep and circadian rhythm abnormalities [16]. Moreover, when compared to wildtype (WT) littermates, Tg2576 mice at age of 12–14 months old were more susceptible to electrically evoked seizures [17], and an increased sensitivity to kindling epileptogenesis [18]. One study reported a high susceptibility to audiogenic-induced seizures in Tg2576 mice that were reduced by passive immunization of an anti-Aβ antibody therapy [19]. Overall, this increased susceptibility to seizures may contribute to a higher mortality rate with Tg2576 mice.
While synaptic loss was absent in Tg2576 mice, changes in synaptic plasticity were reported with impaired LTP measured in the dentate gyrus and CA1 region of hippocampus [20]. In the hippocampus and cortex of aged Tg2576 mice, reduced cholinergic receptor binding and decreased choline uptake were observed supporting cholinergic dysfunction [21, 22]. There were dystrophic cholinergic fibers associated with amyloid plaques [23, 24]. Moreover, cortical neurons derived from Tg2576 mice were found with an impaired retrograde trafficking of BDNF leading to cholinergic degeneration [25, 26].
Besides the absence of a widespread cell loss, Tg2576 mice did not show any build-up of neurofibrillary tangles either [27]. In general, the Tg2576 mouse model is considered as a modest neurodegenerative model possibly due to the promoter used in over-expressing APP [28]. This model was noted with high lethality in certain genetic backgrounds and males tended to be aggressive that required single housing [9]. However, this model is reasonably suited for understanding the pathogenic processes of amyloid in AD [29].
TgCRND8TgCRND8 mice encode a double mutant human APP 695 (the Swedish mutation KM670/671 NL and the Indiana mutation V717F) under the hamster prion protein promoter (PrP) with 5-fold of human APP over-expression [27]. Aβ40 levels were stabilized between 4–10 weeks of age whereas Aβ42 increased slowly between 4–8 weeks with a potent increase at 10 weeks of age [27]. At 2–3 months of age, amyloid deposits were seen in the cerebral cortex. Two months later, dense-core plaques and neuritic pathology began to show in the brain regions of hippocampus, midbrain, brainstem and cerebellum [30]. Interestingly, while TgCRND8 males and females exhibited equivalent Aβ pathologies at 2–8 months of age, females displayed learning and memory deficits much earlier than males [31].
It was reported that TgCRND8 mice manifested with learning impairment detected by the Morris water maze studies as early as 11-week of age that was offset by immunization against Aβ42 [32]. Sex-dimorphic behavioral deficits were described as well. For example, female TgCRND8 mice at 4 months of age had learning and memory deficits detected by the novel object recognition tests [33]. In addition, others described that TgCRND8 males at 5 months of age compensated for Aβ-associated stereotypic behaviors such as hyperactive tight cycling by alternating navigational search strategies and increasingly productive spatial search strategies while females failed to do so [31]. Moreover, sleep-wake cycle dysfunction associated with amyloidosis was reported in male TgCRND8 mice with reduced REM and NREM sleeps at 3–8 months of age, as well as decreased NREM sleep only at 11 months of age [34].
Besides amyloid pathology and behavior impairment, metabolic disturbance was reported in TgCRND8 mice using 1H NMR spectroscopy methods [35]. It was found that levels of N-acetylaspartate (NAA) were reduced in brain regions such as hippocampus and cortex even in young Tg mice (2–3 months old) compared to wt controls prior to any detectable pathological changes, and levels of myo-inositol were decreased in cortical brain regions of young Tg mice but increased in older (12–13 months old) Tg mice [35]. NAA is a metabolite considered to reflect neuronal mitochondrial function [36, 37]. Prior studies implicated a correlation between reduced NAA levels with brain pathology and disease progression in AD patients [38]. It has been found that a decrease in NAA and an increase in myo-inositol both occurred during neuronal cell loss or dysfunction and associated gliosis [39, 40]. These observed changes in NAA and myo-inositol levels suggested underlying neuronal dysfunction or cell loss with associated gliosis [35].
In general, the TgCRND8 mouse model is considered an aggressive model of brain amyloid deposit without tangle pathology. It should be noted that prior report described differences in survival rates and amyloid burden based on different genetic backgrounds in this mouse model with a noticeably shortened life span and a decreased survival rate of TgCRND8 mice with a B6 background but an improved survival rate with less amyloid burden in mice with an A/J inbred background [41]. These findings implicate a layer of complexity with genetic heterogeneity of AD mouse models which could potentially contribute to reported variabilities in AD-related phenotypes and findings in some AD mouse models.
Tau Tg mice PS19This mouse model carries human MAPT (1N4R) tau P301S mutation, under the mouse Prp promoter. This model recapitulates many of the major phenotypes of AD, such as neurofibrillary tangles, synaptic dysfunction, cognitive impairment, and neuronal loss. In this model, human tau expression levels were 5 times that of endogenous mouse tau [42]. There are two primary strains of PS19 mice, namely the mixed background B5;C3 mice and the mixed mice backcrossed with C57BL/6J mice to create a congenic background.
The mixed PS19 mouse model demonstrated pathological tau seeding as early as 1.5 months of age. Tau pathology was developed along neural networks, suggesting tau spread in a prion-like mechanism through neural connections [43]. The initial neuropathological manifestation also included gliosis and impaired synaptic function, followed by synaptic loss. Axonal dysfunction was seen with compromised ER transport as early as 3 months in PS19 mice. At 4 months of age, PS19 mice were positive for neuro-inflammatory markers. These changes occurred prior to the development of tau pathology [42].
The transgenic mice developed neurofibrillary tangles at 6 months of age, and hippocampal and entorhinal cortical atrophy at 9–12 months of age. Specifically, at 12 months of age, there was a 20% reduction in cerebral cortical volume, and a 45% reduction in hippocampus volume. The median life expectancy of PS19 mice was 9 months with 80% of mice dying by 12 months [42]. In a separate study, it was found that female PS19 mice had a significantly higher survival rate compared to male PS19 mice, 90% versus 32% of survival rate at 12 months of age [44]. Interestingly, when 2 months old PS19 mice were immuno-suppressed with FK506 treatment, the survival rate of mice at 12 months of age was increased from 20 to 60% with an associated decrease in neuro-inflammatory markers, neuronal loss, and insoluble hyper-phosphorylated tau. These findings suggest that irregular microglial activation in PS19 mice may exacerbate the effects of tau pathologies and thereby contribute to disease progression [42]. PS19 mice exhibited impairment in memory and learning abilities as early as 3 months of age, before much of the pathology appeared. Mice soon developed limb weakness and later paralysis by 7 months of age [42]. In the Morris water maze test, PS19 mice spent a much longer time finding the invisible platform [45].
The congenic line demonstrates less variability in disease progression, developing neurofibrillary tangles at 6 months and neuronal loss at 9 months of age [46]. They have a median lifespan of 11 to 15 months. Congenic transgenic mice showed a significantly increased tendency to go into and spend time on open arms, suggesting hyper-activities in these mice. It was also reported that PS19 mice showed significantly lower anti-nociceptive responses, with lower thresholds in the hot plate test and lower startle amplitudes in the pre-pulse inhibition test. However, PS19 mice at 3 months of age demonstrated no differences in the tendency to fall when compared to wt counterparts as measured in the rotarod test, suggesting a relatively functional motor tract of these mice at early ages [45].
While the PS19 mouse model simulates many of the AD phenotypes, it does not manifest amyloid pathology. With the high mortality rate in early ages, it is challenging to study tau pathology in this mouse model in later time points. Despite the drawbacks, this model has been used to test novel drug candidates targeting tau pathology, such as microtubule stabilizers like Epothilone D, or lead compounds that reduce tau hyperphosphorylation [47, 48].
rTG4510Similar to the PS19 model, the rTG4510 model over-expressed a human frontotemporal dementia (FTD)-associated MAPT tau mutation. Instead of the P301S tau mutation in PS19, rTG4510 over-expressed the P301L tau mutation with an accelerated formation of a unique 64 kDa hyper-phosphorylated 4R0N isoform of tau. The mouse model was generated by crossing a responder line containing human MAPT P301L cDNA, and a separate line with the tetracycline-controlled trans-activator (tTA) allele under control of the forebrain-specific CaMKIIα promoter. Therefore, human mutant tau transgene expression was largely limited to the hippocampus and neocortex, with associated deficits in hippocampal related activities. It was referred to as the “regulatable” TG4510 as transgene expression can be regulated through doxycycline chow feeding, which provided a temporal control of mutant tau transgene expression [49, 50].
rTg4510 mice expressed 13 times higher of transgenic tau levels compared to endogenous mouse tau levels, forming pre-tangles at 2.5 months of age, and argyrophilic tangle-like inclusions by 4–5.5 months of age in the cortex and the hippocampus [49, 50]. Interestingly, female mice manifested earlier but more severe tau pathology compared to male mice, with significantly increased levels of hyperphosphorylated tau at 5.5 months of age, despite no sex differences in levels of tau transgene expression [51].
A rapid neuronal loss was seen along with tangle formation by 5.5 months of age, and ~60% of decrease in hippocampal CA1 neurons and cortical cell loss occurred at 8.5 months of age [49, 50, 52]. With the progression of tau pathology, axonal degeneration as well as demyelination and impaired white matter integrity were noted by electron microscopy studies [53, 54]. Notably, when suppressing transgenic tau expression through doxycycline chow feeding for 6–8 weeks, CA1 neuronal loss was stabilized, and brain volume loss was prevented after 5.5 to 9 months of feeding. Halting transgenic tau expression at 2.5 months of age stopped the progression of tangle formation and neuronal loss. However, if tau suppression was initiated at 4 months or older, tangle formation proceeded but neuronal loss was prevented suggesting that NFTs may not directly cause neuronal death [50]. It was also found that PS19 mice at 5 months of age had a reduced synchronization of excitatory neurons, which impeded downstream neuronal depolarization and firing. Specifically, membrane potential oscillations during the slow-wave sleep were slowed with altered firing patterns. While few neurons contained NFTs at this age, young mice still experienced spatial memory deficits, indicating that soluble tau may be a primary source of synapse damage, supporting the conclusions from prior studies [55].
Behavioral studies indicated an age-dependent decline in spatial memory function as detected by the Morris water maze tests, severely compromised in rTg4510 mice by 4 months of age. At 7 to 9.5 months of age, Tg mice demonstrated random swimming, with only 25% of time spent in the target quadrant [
留言 (0)