
記住我


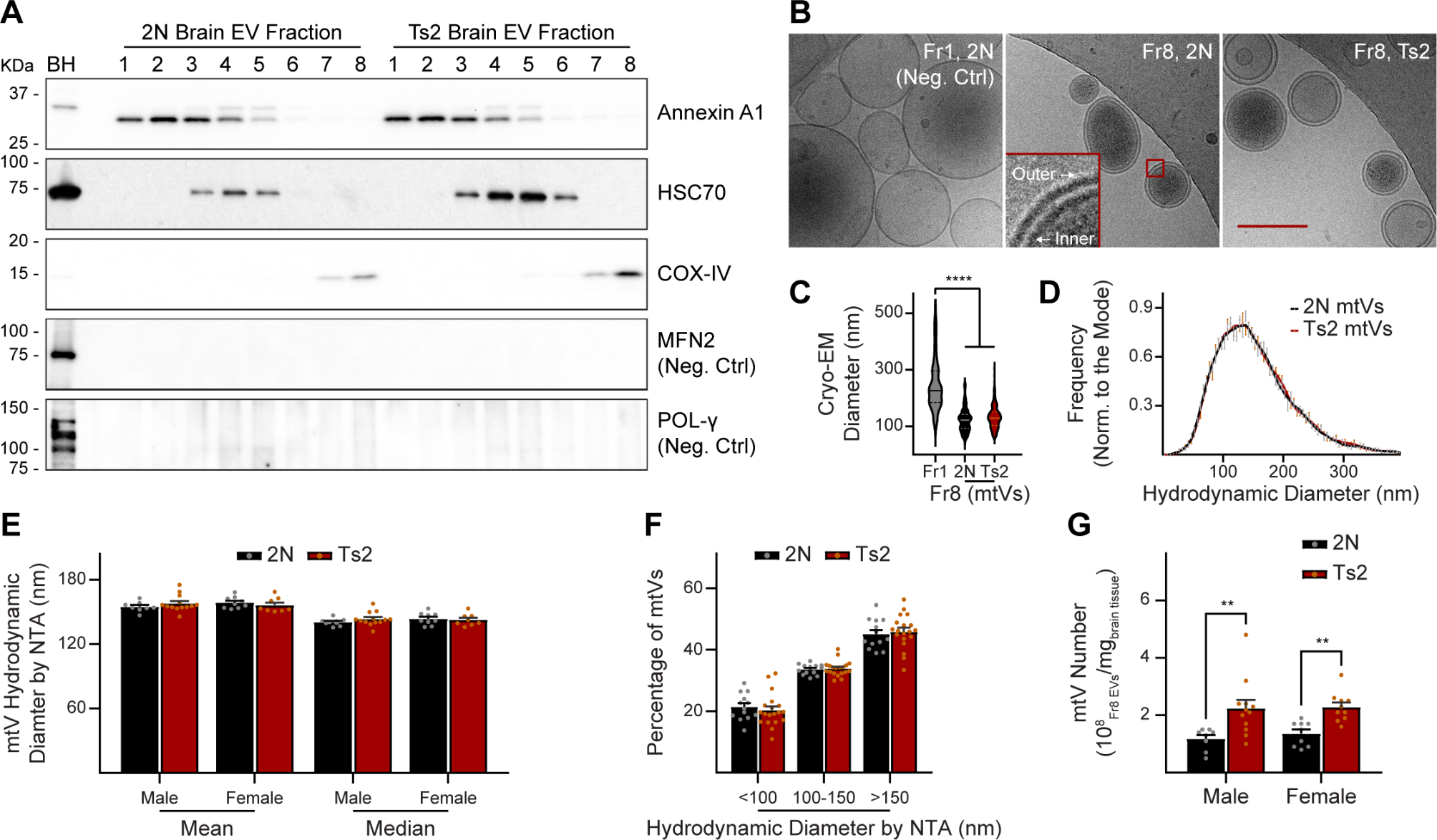

Heterozygous male and female APPSWEPS1L166P (APPPS1-21) mice were maintained on a C57BL/6J background. Heterozygous pups were generated using breeding pairs of (APPPS1-21 Tg male) or (APPPS1-21 Tg female) or (APPPS1-21 dTg x C57BL/6J nonTg). Mice were housed in the University of Chicago Animal Resources Center (ARC) facility under stable housing conditions with a sterile micro-isolator cage (specific-pathogen-free SPF condition). Mice received ad libitum food (Teklad Global, #2918) and water. All experimental procedures were performed following the approved Animal Care and Use Protocols (ACUP) by the Institutional Animal Care and Use Committee (IACUC) of the University of Chicago.
Male and female APPPS1-21 mice were assigned to either vehicle or GV-971 groups in a staggered but randomized manner. A stock solution of three different dosages of GV-971 (40 mg/kg, 80 mg/kg, and 160 mg/kg) was prepared every week using autoclaved ARC drinking water. Using a 24-gauge 1-inch gavage needle (1.25 mm ball), we orally gavaged 8-week-old APPPS1-21 mice with 200 μl of GV-971 (40 mg/kg, 80 mg/kg, 160 mg/kg) every day for one month. Daily weekday/weekend gavages were performed by JM, ensuring that any handling-related distress was avoided in all mice. Also, to nullify any unavoidable handling-related stress effect, control groups of male and female mice received a daily gavage of 200 μl of autoclaved ARC drinking water. In this study, attention was given to mice developing any significant changes in health. Notably, there were no mice in the study that requires removal due to health issues. Mice were sacrificed at three months of age using the IACUC-approved euthanasia protocol.
WashU 5XFADHeterozygous male and female B6.Cg-Tg (APPSwFf1Lon, PSEN1*M146L*L286V) 6799Vas/Mmjax (5XFAD) mice [26]were purchased from Jackson Laboratory (MMRRC Strain #034840-JAX). All mice are on the C57BL/6 background. Heterozygous pups were generated using breeding pairs of (5XFAD Tg males x C57BL/6J nonTg females) or C57BL/6J nonTg males x 5XFAD females). Mice were housed under normal 12-h light/dark cycles under stable housing conditions with a sterile micro-isolator cage (specific-pathogen-free SPF condition). Mice received ad libitum food (PicoLab Rodent Diet 20; #5053) and water. All animal studies were approved by the Animals Studies Committee at Washington University School of Medicine in St. Louis.
Male and female 5XFAD mice were assigned to either vehicle or GV-971 groups in a randomized manner. A stock solution of GV-971 was prepared every week using autoclaved drinking water. Using an 18-gauge 2-inch gavage needle (1.25 mm ball), 7-month-old mice were orally gavaged every day with 100 mg/kg of GV-971 or vehicle for 2 months. Daily weekday/weekend gavages were performed by MEB, ensuring that any handling-related distress was avoided in all mice. To nullify any unavoidable handling-related stress, vehicle mice received a daily gavage of autoclaved drinking water. Mice were sacrificed at 9 months of age using IACUC-approved euthanasia protocol.
Necropsy and tissue harvestingU. Chicago APPPS1-21Perfusion and tissue harvesting were carried out as described in [19]. On the day of sacrifice, mice were sedated using ketamine/xylazine as per the approved ACUP. The blood samples were collected by cardiac puncture using a 25-gauge needle and stored at 40C, in sodium citrate buffer tubes (BD Vacutainer; #363083) prior to the centrifugation. Transcardial perfusion was performed using physiological pH cold saline through the left-ventricle and simultaneously clamping the descending aorta. Harvested brains were dissected into two hemispheres (one hemisphere was post-fixed with 4% paraformaldehyde and the other was frozen for RNA extractions and MSD biochemical analysis). Cecum was collected from the non-perfused lower body and stored at -800C until the use. Plasma samples were separated by centrifugation at 2,000 rpm for 10 min at 40C and stored at -800C.
WashU 5XFADPerfusion and tissue harvesting was carried out as described in [27]. On the day of sacrifice, mice were sacrificed by intraperitoneal injection of pentobarbital (200 mg/kg). Blood samples were collected in EDTA-treated tubes before cardiac perfusion with 3 U/ml heparin in cold Dulbecco’s PBS. Blood samples were spun down (10 min, 2,000 × g, 4 °C), and blood plasma was collected. Transcardial perfusion was performed using physiological pH cold saline with 3 U/ml heparin. Brains were extracted and cut into two hemispheres. The left hemisphere was collected for immunostaining and immerse-fixed. The right hemisphere was dissected to isolate the hippocampus and cortex for biochemical analysis, and the tissue was kept at − 80 °C until analyzed. Fecal samples were collected using the clean catch method into 1.7 mL Eppendorf tubes and stored at -800C until the use.
ImmunohistochemistryU. Chicago APPPS1-21Histology was performed as per the established procedure [19, 20, 28]. Briefly, extracted hemispheres were post-fixed using 4% paraformaldehyde for 24 h then, brains were transferred into 30% sucrose. Leica microtome (Leica; #SM210R,) was used to cut 40 µm thick coronal brain sections (from the beginning of the olfactory bulb till the end of hippocampal level sections) and stored in cryoprotectant solution. On the day of staining, a total of 6 level-matched sections at an equidistant interval of 480 µm were selected from each mouse for free-floating staining. Tissue slices were washed and blocked with serum-blocking solution for 1 h. Primary antibody specific for human amyloid (3D6, in house antibody, 1:10000) was used for 48 h incubation, and secondary antibody (Donkey-anti-mouse 488, 1:500, Thermofisher; # A-21202,) incubation for one hour. The sections were mounted on glass slides and then coverslipped using amount mounting media (ThermoFisher; # F4680,) prior to submission to the University of Chicago microscope core facility for full slide scanning. Immunofluorescence staining using primary antibodies (anti-amyloid (mouse-anti-Aβ, 3D6, 1:10000), homeostatic microglia (Rabbit-anti- P2ry12: Sigma-Aldrich; # HPA014518, 1:1000) and neurodegenerative-type microglia (Rat anti-mDectin-1: InvivoGen; # mabg-mdect, 1:250) markers) and respective secondary antibodies (Donkey-anti-mouse 488, Goat-anti-rat 555, Donkey-anti-rabbit 647) were performed to investigate the status of plaque-localized microglial cells. We used three level-matched sections at an equidistance of 960 µm to investigate the plaque-localized microglia status as described below.
WashU 5XFADHistology was performed as established in [27]. Briefly, extracted hemispheres were post-fixed using 4% paraformaldehyde for 48 h, then, brains were transferred into 30% sucrose. Leica microtome (Leica; #SM210R) was used to cut 30 µm thick coronal brain sections (from the beginning of the olfactory bulb till the end of hippocampal level sections) and stored in cryoprotectant solution. A total of 3 level-matched sections at an equidistant interval of 360 µm were selected from each mouse for free-floating staining. For Aβ plaque analysis, sections were washed three times in TBS for 5 min and blocked in 0.3% hydrogen peroxide for 10 min. After washing, sections were blocked in 3% milk in TBS with 0.25% Triton X-100 (TBSX) for 30 min. The sections were incubated in primary antibody (HJ3.4 biotinylated, anti-Aβ1–13, mouse monoclonal, 2 µg/ml generated in-house) overnight at 4 °C. Sections were incubated in ABC Elite solution (VectaStain; #PK-6100) for 1 h, prepared following the manufacturer’s instructions. Sections were developed in DAB solution (Sigma; #D5905), washed, and mounted on slides and cover slipped with Cytoseal 60 (Thermo Fisher Scientific; #8310). Immunofluorescence staining using primary antibodies (anti-amyloid (mouse-anti-amyloid, HJ3.4 biotinylated, anti-Aβ1–13, mouse monoclonal, 2 µg/ml, X-34 1;1000 Sigma; #SML1954), homeostatic microglia (Rabbit-anti- P2ry12: Biolegend; #S16007D, 1:100) and neurodegenerative-type microglia (Rat anti-mDectin-1(Clec7a): InvivoGen; #mabg-mdect, 1:100) markers), general microglia marker (Iba-1 Wako; 011–27991, 1:1000), astrocyte activation (GFAP Abcam; #ab134436, 1:1000) and respective secondary antibodies (Donkey-anti-rabbit 488 (#711–545-152), Donkey-anti-rat 594 (#712–585-150), Donkey-anti-chicken 647 (#703–605-155), Jackson ImmunoResearch) incubations were performed to investigate the status of plaque-localized microglia and astrocytes. Lipofuscin was quenched with 1X TrueBlack (Biotium; 23007) and washed once in PBS. Sections were mounted and sealed in ProLong Gold anti-fade (Thermo Fisher Scientific;#P36930). We used three level-matched sections at an equidistance of 360 µm to investigate the plaque-localized microglia and astrocytes status as described below.
Amyloid-β burden measuresU. Chicago APPPS1-21Amyloid quantification was performed as published previously [19, 20, 28]. In brief, slides were scanned using a slide scanner under a magnification of 20x to prepare a 3D Z-stack of each slide by the microscope core facility. Threshold-based analysis for Aβ burden was performed on these images using Fiji Image-J. The sections were converted into 8-bit images, followed by a selection of cerebral cortex following tissue landmarks. An appropriate threshold number (based on the preliminary analysis avoiding floor/ceiling effects) was applied consistently to highlight the majority of amyloid plaques ensuring no artifacts were incorporated in the quantification. Fill holes and watershed plugins were applied. Particles were analyzed using 10-400pixel2 size and 0.4–1.00 circularity criteria for each section. Collated numbers were collected to calculate the mean burden and mean particle size and compared between groups using GraphPad Prism software (Prism 7, version 7.0e, 2018).
WashU 5XFADImages were obtained from an average of 3 sections per mouse for IHC and IF. For IHC stains, slides were scanned on the NanoZoomer 2.0-HT system (Hamamatsu Photonics) at 20x. Images were further processed using NDP viewing software (Hamamatsu Photonics) and Fiji software version 1.51 (National Institutes of Health). The sections were converted into 8-bit images followed by a selection of cerebral cortex and hippocampus using tissue landmarks. An appropriate threshold number (based on the preliminary analysis avoiding floor/ceiling effects) was applied consistently to highlight the majority of amyloid plaques ensuring no artifacts were incorporated in the quantification. Particles were analyzed and collated numbers were collected to calculate the mean amyloid burden between groups using GraphPad Prism software. For IF (Iba1 and GFAP) stains and slides were scanned on Leica Thunder imager 3D assay at 20x. Images were further processed using Fiji software as previously described.
Microglia (Clec7a+/P2ry12+ plaque-localized microglia) quantificationU. Chicago APPPS1-21Immunofluorescence slides stained with markers for 3D6 (FITC), Clec7a (Cy5), and P2ry12 (Cy3) were scanned using Leica SP8 laser scanning confocal microscope under 63x/1.4 UV oil objective. 3D-Z stacks, 0.35 µm step increments in the z-plane, were prepared for 10 plaques-containing microenvironments per case. The images were then imported into Fiji Image-J for individual channel separation, and maximum-intensity projections, followed by manual counts of Clec7a+ and P2ry12+ microglia numbers in each compressed image containing Aβ plaque. The average number of Clec7a+ microglia and P2ry12+ microglia per image (0.02mm2 area) was generated and plotted using GraphPad Prism (Prism 7, version 7.0e, 2018).
WashU 5XFADImmunofluorescence slides were stained with markers for Ibal-1 (Alexa 488), Clec7a or P2ry12 (Alexa 594), and HJ3.4 (Alexa 647) were scanned using Leica SP5 laser scanning confocal microscope under 20x objective. Three 3D-Z stacks, 2 µm step increments in the z-plane, were acquired from 3 cortical tissues per mouse, 9 images total. Quantification of confocal images for Iba1/plaque, Clec7a/ plaque, and P2ry12/plaque was performed on a semi-automated platform using MATLAB and Imaris 9.3.1 software to create surfaces of each stain. The Aβ plaque surfaces were then extended 3-15 μm around and the number of counter stain surfaces within the plaque perimeter was quantified and plotted using GraphPad Prism (Prism 9.4.1).
Cerebral cortex RNA extractionU. Chicago APPPS1-21Total RNA was isolated from the dorsal cerebral cortex similar to our previous work [20]. Briefly, the dorsal cerebral cortex was homogenized with TRIzol reagent and RNA was extracted. The quality of the total RNA was evaluated using the Agilent Bioanalyzer. RNA-seq library preparations and illumine HiSeq4000 were performed by the University of Chicago Genomics Core facility similar to sequencing generated previously [20]. The data files were collected in FASTQ format for the bioinformatic analysis.
WashU 5XFADTotal RNA was isolated from whole cerebral cortex using the Trizol method followed by RNeasy Mini Kit (QIAGEN; catalog no. 74104) and prepared cDNA with the High-Capacity RNA-to-cDNA kit (Applied Biosystems; #4388950) following the manufacturer’s instructions. cDNA was further purified using a QIAquick PCR purification kit following the manufacturer’s instructions (QIAGEN; #28104).
U. Chicago APPPS1-21 RNA-seq bioinformatics analysisThe quality of RNA reads, in FASTQ format, was evaluated using FastQC [29] and similar to our published work [19]. In brief, adapters were trimmed, and reads of poor quality or aligning to rRNA sequences were filtered using Trim Galore (http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/). The later reads were then aligned to the mouse genome (mm10) using STAR [30]. Read counts for each gene were calculated using HTSeq-Counts [29] in conjunction with a gene annotation file for mm10 obtained from Ensembl (http://useast.ensembl.org/index.html). We generated a comprehensive quality control report using MultiQC [30]. DEseq2 [31] was used to determine differential expression. The cut-off for determining significant DEGs was a P < 0.01. GO analysis and identification of DEGs belonging to specific pathways were performed using Metascape [32]. We also utilized Cytoscape to analyze string networks and especially to investigate the immune pathway-related targets. Gene set enrichment analysis (GSEA) was also utilized to compare our cerebral cortex total RNAseq-generated transcriptome data with available online deposited gene enrichment datasets. Data generated from these different platforms were utilized to generate Figs. 7 and 8.
U. Chicago APPPS1-21 reverse transcription quantitative PCRWe used 1 μg of RNA for cDNA synthesis using the SuperScript IV VILO Master Mix with exDNase Enzyme (ThermoFisher; #11766050) according to the kit instructions. After synthesis, cDNA was diluted 40 times in Nuclease-free water (Invitrogen; #AM9938). Primers used for the detection of the signal were designed either by using established ones in previously published methods or using Primer BLAST tool (NCBI distribution) and checked for dimer formation in Multiple Primer Analyzer software (ThermoFisher). cDNA were subject to qPCR using PowerUp SYBR Green Master Mix (Applied Biosystems by Life Technologies; #A25742,). The volume of each reaction was 10 μL, which contained 5μL of PowerUP SYBR Green Master Mix (2x), 0.5 μL of forward and reverse primer mixture (in concentration of 5 μM), 0.4 μL of Nuclease-free water, and 4 μL of cDNA added to wells individually. The reactions were run on MicroAmp Fast Optical 96-Well Reaction Plate with Barcode (0.1 mL, Applied Biosystems by Life Technologies;# 4346906) with MicroAmp Optical Adhesive Film (Applied Biosystems by Life Technologies; 4311971,) on QuantStudio 3 Real-Time PCR System (ThermoFisher; #A28567,). Amplification was performed starting with 3 min hold at 95 °C to activate the enzyme. Next, the template was denaturated at 95 °C for 20 s, then annealed at 60 °C for 20 s, which was followed by extension and data acquisition at 72 °C for 20 s.). In brief, the data was normalized by evaluation of Ct mean of a housekeeping gene (Cyc1) for each sample. Cyc1 was the most stable housekeeping gene, hence only this gene was included in analysis. The expression levels were then calculated according to the ΔΔCt method. To assess the statistical differences between gene expressions two-way ANOVA was performed. A p-value of < 0.05 was considered statistically significant.
WashU 5XFAD Fluidigm qPCRGene expression analysis was performed using microarray in collaboration with the Genome Technology Access Core at Washington University. Using TaqMan probes, the relative gene expression was quantitatively measured using Fluidigm Biomark HD with integrated fluidic circuits. The data was normalized by evaluation of geometricCt mean of housekeeping genes (GAPDH and β-actin) for each sample. The expression levels were then calculated according to the ΔΔCt method. To assess the statistical differences between gene expressions a t-test was performed. A p-value of < 0.05 was considered statistically significant.
U. Chicago APPPS1-21brain protein extractionTo quantify soluble and insoluble Aβ levels, we chose the ventral half of each frozen brain. Briefly, the -80 °C stored frozen brains were dissected into dorsal and ventral halves on dry ice. The ventral halves were weighed and homogenized with 5X (w/v) volume tris-buffered saline (TBS) containing 1X halt protease inhibitor cocktail (Thermo Fisher Scientific) and 5 mM EDTA. After sonication, the homogenized samples were subject to ultracentifugation at 100,000xg for 60 min at 4 °C. The supernatant fraction was collected to detect TBS soluble Aβ levels. The remaining pellet fraction was further extracted in 10X (w/v) volume of 70% formic acid (FA) followed by homogenization. The homogenized samples were then subject to ultracentrifugation at 100,000xg for 60 min at 4 °C to collect the supernatant fraction that contains TBS-insoluble and FA soluble Aβ levels. The supernatants were frozen immediately and shipped on dry ice to the Harvard University for MesoScale Aβ analysis.
U. Chicago APPPS1-21 MesoScale Aβ analysisLevels of Aβ peptides were analyzed through protocols previously reported [33]. Specifically, the assay was performed on an electrochemi-luminescence-based multi-array method using the Quickplex SQ 120 system from MSD Meso Scale Diagnostics LLC. The MesoScale Aβ 4G8 kits were utilized to detect Aβ peptides in a 96-well-based assay. First, 96-well plates were blocked with diluents provided by the manufacturer with shaking for 1 h at room temperature (RT). The experimental samples and MesoScale protein standards were resuspended in the manufacturer-supplied detection antibodies. The mixed solutions were placed on a shaker for 2 h at RT, followed by washing and adding of the reading buffer. The electrochemiluminescence signals were captured and signals were obtained for all samples and standard proteins, and the sample Aβ levels were analyzed using the MesoScale protein standards.
U. Chicago APPPS1-21Cecal bacterial DNA extraction and microbiome analysisWe used 50-100 mg of cecal content to extract microbial DNA. DNA extractions were performed using Qiagen DNeasy PowerSoil Pro kit following the manufacturer’s protocol (Qiagen; # 47,016) by IQW. Purified DNA was submitted to the Argonne National Laboratories for 16 s rRNA amplicon sequencing (Illumin MiSeq) under EGC supervision. For sequencing analysis, Earth Microbiome Project raw sequences were imported into Qiime2 [34] and analyses were performed by HBD. We used Dada2 to demultiplex [33]. Followed by quality control, sequences were aligned using mafft [35] and a phylogenetic tree was constructed using fasttree [36]. Sampling depth was rarefied at 9000sequences per sample for combined comparison irrespective of sexes and separately at 14400sequences per sample for male groups and 9000sequences per sample for female groups to maximize depth while prioritizing equal retention of samples across groups [37]. Alpha diversity and β diversity were calculated as per established protocols [38,39,40]. Taxonomy was compiled using the classify-sklearn plugin with greengenes Greengenes 13_8 99% OTUs pre-trained Naïve Bayes classifier [41,42,43]. ANCOM (analysis of comparison of microbiome) was performed using ANCOM plugin to evaluate differentially abundant taxa at species level (L7) between male groups and female groups.
WashU 5XFAD fecal bacterial DNA extraction and microbiome analysisWe used 20-50 mg of fecal content to extract microbial DNA. DNA extractions were performed using Qiagen DNeasy PowerSoil Pro kit following the manufacturer’s protocol (Qiagen; # 47016) by MEB. Purified DNA was submitted to the Washington University McDonnell Genome Institute under MLL supervision. Seven PCR amplicons representative of all nine 16S variable regions using the primers indicated in Table 1 were generated using the Fluidigm Access Array System. Reaction mixture components included 10X Fast Start High Fidelity buffer without MgCl, 25 mM MgCl, dimethyl sulfoxide, 10 mM PCR Grade Nucleotide Mix, 0.05 U/µL of 5U/µL FastStart High Fidelity Enzyme Blend, 20X Access Array Loading Reagent, 1 µL DNA, and molecular grade water. The BioMark HD system from Fluidigm was employed for PCR amplification. Reaction products were indexed with unique 10 base pair sequences via 7 rounds of PCR in order to combine each index sequence. 48 sample libraries were constructed via sample pooling and bead purification used for cleaning. Illumina MiSeq sequencer (2 × 150 base pair kit) was used for library sequencing. Amplification and sequencing were performed at the Genome Technology Access Center at the McDonnell Genome Institute at Washington University in St. Louis. Demultiplexed reads from the 7 amplicons were analyzed using the MVRSION pipeline [44] to generate a list of microbial species with their corresponding number of reads for each sample. Default parameters were employed for the MVRSION analysis in conjunction with the Silva 16S database and data was rarefied to a depth of 11,000 for the female group and 22,000 for male groups. Diversity analysis was run for the following sample groupings: sex and treatment. The taxonomic classification results of MVRSION were post-processed via QIIME for the core set of diversity analyses, including alpha and beta diversity, alpha rarefaction, and group significance. Additional alpha diversity analysis employed QIIME2.
Table 1 GV-971 significantly alters microbiome bacterial species in male mice treated with 160 mg/kg compared with vehiclePrimers sequences associated with the seven PCR amplicons covering 9 variable regions in the bacterial 16S rRNA gene are listed below.
Name
Sequence
V1-V2_F
TCGTCGGCAGCGTCAGAGTTTGATCCTGGCTCAG
V2_F
TCGTCGGCAGCGTCAGYGGCGIACGGGTGAGTAA
V3_2_F
TCGTCGGCAGCGTCCCTACGGGAGGCAGCAG
V4_F
TCGTCGGCAGCGTCGTGCCAGCMGCCGCGGTAA
V5-V6_F
TCGTCGGCAGCGTCAGGATTAGATACCCTGGTA
V6_1_F
TCGTCGGCAGCGTCAAACTCAAAKGAATTGACGG
V7-V8_F
TCGTCGGCAGCGTCGYAACGAGCGCAACCC
V1-V2_R
GTCTCGTGGGCTCGGTGCTGCCTCCCGTAGGAGT
V2_R
GTCTCGTGGGCTCGGCYIACTGCTGCCTCCCGTAG
V3_2_R
GTCTCGTGGGCTCGGGTATTACCGCGGCTGCTGG
V4_R
GTCTCGTGGGCTCGGGGACTACHVGGGTWTCTAAT
V5-V6_R
GTCTCGTGGGCTCGGCRRCACGAGCTGACGAC
V6_1_R
GTCTCGTGGGCTCGGACGAGCTGACGACARCCATG
V7-V8_R
GTCTCGTGGGCTCGGGACGGGCGGTGWGTRC
WashU and U. Chicago Luminex cytokine/chemokine arrayMouse serum and brain tissue lysate samples were thawed on ice, centrifuged at 15,000 rcf for 10 min at 4C to remove particulates and aggregates, then 25 uL of sample or prepared kit standard was added to each well (in duplicate) of a 96 well plate containing premixed beads and assay buffer. The standard for the tissue lysates was prepared with the lysis buffer (Invitrogen ProcartaPlex cell lysis buffer). The bead-based multiplex immunoassay was performed according to the manufacturer’s instructions (ThermoFisher Procartaplex Mouse Cytokine/Chemokine Panel 1A 36plex; # EPXR360-26092–901). The panel probed for the following chemokines and cytokines: ENA-78 (CXCL5), Eotaxin (CCL11), GRO alpha (CXCL1), IP-10 (CXCL10), MCP-1 (CCL2), MIP-1 alpha (CCL3), MIP-1 beta (CCL4), MIP-2 alpha (CXCL2), RANTES (CCL5) Cytokines: G-CSF (CSF-3), GM-CSF, IFN alpha, IFN gamma, IL-1 alpha, IL-1 beta, IL-2, IL-3, IL-4, IL-5, IL-6, IL-9, IL-10, IL-12p70, IL-13, IL-15/IL-15R, IL-17A (CTLA-8), IL-18, IL-22, IL-23, IL-27, IL-28, IL-31, LIF, MCP-3 (CCL7), M-CSF, TNF alpha. The plate was incubated on a shaker at 700 rpm for 2 h at room temperature, washed on a hand-held magnet, then the detection antibody was added for 30 min. After washing the unbound detection antibody away from the beads, the streptavidin phycoerythrin (SA-PE) reagent was added to each well and the plate shaken for 30 min. A final wash was performed then the beads were read for MFI using a FLEXMAP3D Luminex (Luminex Corp, Austin, TX) machine. MilliporeSigma Belysa v.1 (Merck EMD Millipore, Billerica, MA) analysis software was used to calculate the pg/ml for each analyte using a 5-parameter logistical curve-fit algorithm.
Metabolite extraction from cecal materialExtraction solvent (80% methanol spiked with internal standards and stored at -80 °C) was added to pre-weighed fecal/cecal samples at a ratio of 100 mg of material/mL of extraction solvent in beadruptor tubes (Fisherbrand; 15–340-154). Samples were homogenized at 4 °C on a Bead Mill 24 Homogenizer (Fisher; 15–340-163), set at 1.6 m/s with 6 thirty-second cycles, 5 s off per cycle. Samples were then centrifuged at -10 °C, 20,000 × g for 15 min and the supernatant was used for subsequent metabolomic analysis.
Metabolite analysis using GC-nCI-MS and PFBBr derivatizationMetabolites were derivatized as described by Haak et al. [26] with the following modifications: the metabolite extract (100μL) was added to 100μL of 100 mM borate buffer (pH 10) (Thermo Fisher, #28341), 400μL of 100 mM pentafluorobenzyl bromide (Millipore Sigma; #90257) in acetonitrile (Fisher; A955-4), and 400μL of n-hexane (Acros Organics; #160780010) in a capped mass spec autosampler vial (Microliter; 09–1200). Samples were heated in a Thermomixer C (Eppendorf) to 65 °C for 1 h while shaking at 1300 rpm. After cooling to RT, samples were centrifuged at 4 °C, 2000 × g for 5 min, allowing phase separation. The hexane phase (100μL) (top layer) was transferred to an autosampler vial containing a glass insert and the vial was sealed. Another 100μL of the hexane phase was diluted with 900μL of n-hexane in an autosampler vial. Concentrated and dilute samples were analyzed using a GC–MS (Agilent 7890A GC system, Agilent 5975C MS detector) operating in negative chemical ionization mode, using a HP-5MSUI column (30 m × 0.25 mm, 0.25 μm; Agilent Technologies; #19091S-433UI), methane as the reagent gas (99.999% pure) and 1μL split injection (1:10 split ratio). Oven ramp parameters: 1 min hold at 60 oC, 25 oC per min up to 300 °C with a 2.5 min hold at 300 °C. Inlet temperature was 280 °C and transfer line was 310 °C. A 10-point calibration curve was prepared with acetate (100 mM), propionate (25 mM), butyrate (12.5 mM), and succinate (50 mM), with 9 subsequent 2x serial dilutions. Data analysis was performed using MassHunter Quantitative Analysis software (version B.10, Agilent Technologies) and confirmed by comparison to authentic standards. Normalized peak areas were calculated by dividing raw peak areas of targeted analytes by averaged raw peak areas of internal standards.
LCMS/MSIndole-containing metabolites, B-vitamins and other targeted metabolites were analyzed by LCMS/MS. The metabolite extract (400μL) was added to pre-labeled microcentrifuge tubes. Samples were dried down completely using a Genevac EZ-2 Elite. Samples were resuspended in 100μL of 50:50 Water: Methanol and added to an Eppendorf thermomixer. C at 4 oC, 1000 rpm for 15 min to resuspend analytes. Samples were then centrifuged at 4 °C, 20,000 × g for 15 min to remove insoluble debris. The supernatant (80μL) was transferred to a fresh, prelabeled MS vial with inserts or 96 deep-well plate (Agilent; #5065–4402). Samples were analyzed on an Agilent 1290 infinity II liquid chromatography system coupled to an Agilent 6470 triple quadrupole mass spectrometer, operating in positive mode, equipped with an Agilent Jet Stream Electrospray Ionization source. Each sample (2μL) was injected into a Acquity UPLC HSS PFP column, 1.8 μm, 2.1 × 100 mm (Waters; 186005967) equipped with a Acquity UPLC HSS PFP VanGuard Precolumn, 100., 1.8 μm, 2.1 mm X 5 mm (Waters; 186005974) at 45 oC. Mobile phase A was 0.35% formic acid in Water and mobile phase B was 0.35% formic acid in 95:5 Acetonitrile:Water. The flow rate was set to 0.5 mL/min starting at 0% B held constant for 3 min, then linearly increased to 50% over 5 min, then linearly increased to 95% B over 1 min, and held at 100% B for the next 3 min. Mobile phase B was then brought back down to 0% over 0.5 min and held at 0% for re equilibration for 2.5 min. The QQQ electrospray conditions were set with capillary voltage at 4 kV, nozzle voltage at 500 V, and Dynamic MRM was used with cycle time of 500 ms. Transitions were monitored in positive mode for 46 analytes (table on next slide). An 11-point calibration curve (ranging from 0.88 nM to 909 μM) was prepared for tryptophan, tyrosine, phenylalanine, serotonin, 5-HIAA, melatonin, tryptamine, kynurenine, kynurenic acid, anthranilic acid, and niacin. Data analysis was performed using MassHunter Quant software (version B.10, Agilent Technologies) and confirmed by comparison with authentic standards. Normalized peak areas were calculated by dividing raw peak areas of targeted analytes by averaged raw peak areas of internal standards.
Bile acid analysisBile acids were analyzed using LCMS. The metabolite extract (75μL) was added to prelabeled mass spectrometry autosampler vials (Microliter; 09–1200) and dried down completely under a nitrogen stream at 30 L/min (top) 1 L/min (bottom) at 30 °C (Biotage SPE Dry 96 Dual; #3579 M). Samples were resuspended in 50:50 Water: Methanol (750μL). Vials were added to a thermomixer C (Eppendorf) to resuspend analytes at 4 °C, 1000 rpm for 15 min with an infinite hold at 4 °C. Samples were then transferred to prelabeled microcentrifuge tubes and centrifuged at 4 °C, 20,000 × g for 15 min to remove insoluble debris. The supernatant (700μL) was transferred to a fresh, prelabeled mass spectrometry autosampler vial. Samples were analyzed on a liquid chromatography system (Agilent 1290 infinity II) coupled to a quadrupole time-of-flight (QTOF) mass spectrometer (Agilent 6546), operating in negative mode, equipped with an Agilent Jet Stream Electrospray Ionization source. The sample (5μL) was injected onto an XBridge© BEH C18 Column (3.5 μm, 2.1 × 100 mm; Waters Corporation, PN) fitted with an XBridge© BEH C18 guard (Waters Corporation, PN) at 45 °C. Elution started with 72% A (Water, 0.1% formic acid) and 28% B (Acetone, 0.1% formic acid) with a flow rate of 0.4 mL/min for 1 min and linearly increased to 33% B over 5 min, then linearly increased to 65% B over 14 min. Then the flow rate was increased to 0.6 mL/min and B was increased to 98% over 0.5 min and these conditions were held constant for 3.5 min. Finally, re-equilibration at a flow rate of 0.4 mL/min of 28% B was performed for 3 min. The electrospray ionization conditions were set with the capillary voltage at 3.5 kV, nozzle voltage at 2 kV, and detection window set to 100–1700 m/z with continuous infusion of a reference mass (Agilent ESI TOF Biopolymer Analysis Reference Mix) for mass calibration. A ten-point calibration curve was used for quantitation. Data analysis was performed using MassHunter Profinder Analysis software (version B.10, Agilent Technologies) and confirmed by comparison with authentic standards. Normalized peak areas were calculated by dividing raw peak areas of targeted analytes by averaged raw peak areas of internal standards.
Statistical analysisWe used GraphPad Prism (version 7.0e) to run statistical analyses. Two-way ANOVA were performed to evaluate different parameters among vehicle vs treatment groups in a sex-specific manner unless otherwise noted. For the 5XFAD study, sexes were separated and student t-test was utilized to compare GV-971 with controls. Statistical P value below 0.05 was considered for significant differences unless otherwise noted. For microbiome data analysis, Kruskal–Wallis (non-parametric) comparisons were performed to compare several diversity-related indices. ANCOM was performed as mentioned above to evaluate the differences at species level between groups. QIIME and QIIME2 was utilized to determine diversity analyses, including alpha and β diversity, alpha rarefaction, and group significance for the 5XFAD microbiome analysis.
留言 (0)