
記住我








This trial is a single-center, prospective, double-blinded, randomized controlled trial (RCT) using a two-arm, parallel group design. The study protocol has been approved by the Ethics Committee of the 920th Hospital of Joint Logistics Support Force and has been registered with the Chinese Clinical Trial Registration Center (ChiCTR2300069458). This trial protocol is produced according to the applicable Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) reporting guidelines [13].
Eligibility CriteriaThe inclusion criteria: (1) age ≥ 18 years old; (2) American Society of Anesthesiologists grade (ASA) I or II; (3) body mass index (BMI) 18–30 kg/m2. The exclusion criteria: (1) TEAS contraindications (scars or skin damage/infection at the stimulation site, electronic devices such as pacemakers or other medical electronic devices implanted in the body); (2) a history of chronic pain; (3) diabetes and/or hypertension (Persistent electrical stimulation may cause severe fluctuations in blood pressure or blood glucose.); (4) confirmed heart/brain/liver/renal with severe dysfunction (Patients who have confirmed heart/brain/liver/renal with severe dysfunction may result in delayed extubation after surgery. Liver/renal with severe dysfunction will affect drug metabolism and may affect the perioperative medication of patients.); (5) a history of acupuncture or TEAS (Patients may be aware of their group assignment.); (6) drug or alcohol abuse/addiction; (7) communication difficulties.
Randomization and BlindingParticipants who meet the screening criteria will be randomized into two balanced groups: TEAS group and sham group. The random allocation sequence will be computer-generated with a 1:1 ratio using IBM SPSS Statistics V25.0.
Allocations of subjects will be kept confidential, unless severe postoperative pain occurs during the trial and cannot be relieved, or the patient requests that the trial be terminated. After unblinding, patient trial treatment will be discontinued, and the clinical information of the unblinded patients will be recorded in detail.
The investigators will be responsible for recruitment and sign informed consent with patients. After successful recruitment, the clinicians will perform TEAS or no TEAS according to randomization. The patients, outcome assessor, and statistician will be blinded. Other clinical staff (surgeons and ward staff) are not aware of the grouping situation.
To maintain blinding, in both group, electrodes will be attached to the same acupoints and then connected with electrical stimulator. Sham group patients will receive sham stimulation by connecting the same acupoints on their limbs. We will inform the patient that due to individual difference, each person may experience the stimulation differently.
Sample SizeAccording to the incidence of chronic pain after thoracoscopic surgery, which is as high as 43.99% [5], the sample size is calculated as a bilateral test and taken α with a value of 0.05, selecting a total of 64 people from each group of 32 as the total sample size can achieve a testing efficiency of 80% and an inter group difference of 30%. Considering a postoperative drop-out rate of 20%, a total of 80 cases will be selected as the sample size for this trial.
Recruitment Strategies and EnrollmentEighty patients who meet the eligibility criteria and sign the informed consent will be recruited at the 920th Hospital of Joint Logistics Support Force. We plan to enroll the first patient on March 20, 2023 and to end on March 20, 2024. All participants will be randomly divided into two groups: an experimental group (TEAS group) and a control group (sham group). The researchers’ screen subjects will be based on established standard treatment plans. Date collection will start from the collection of baseline data until the end of follow-up.
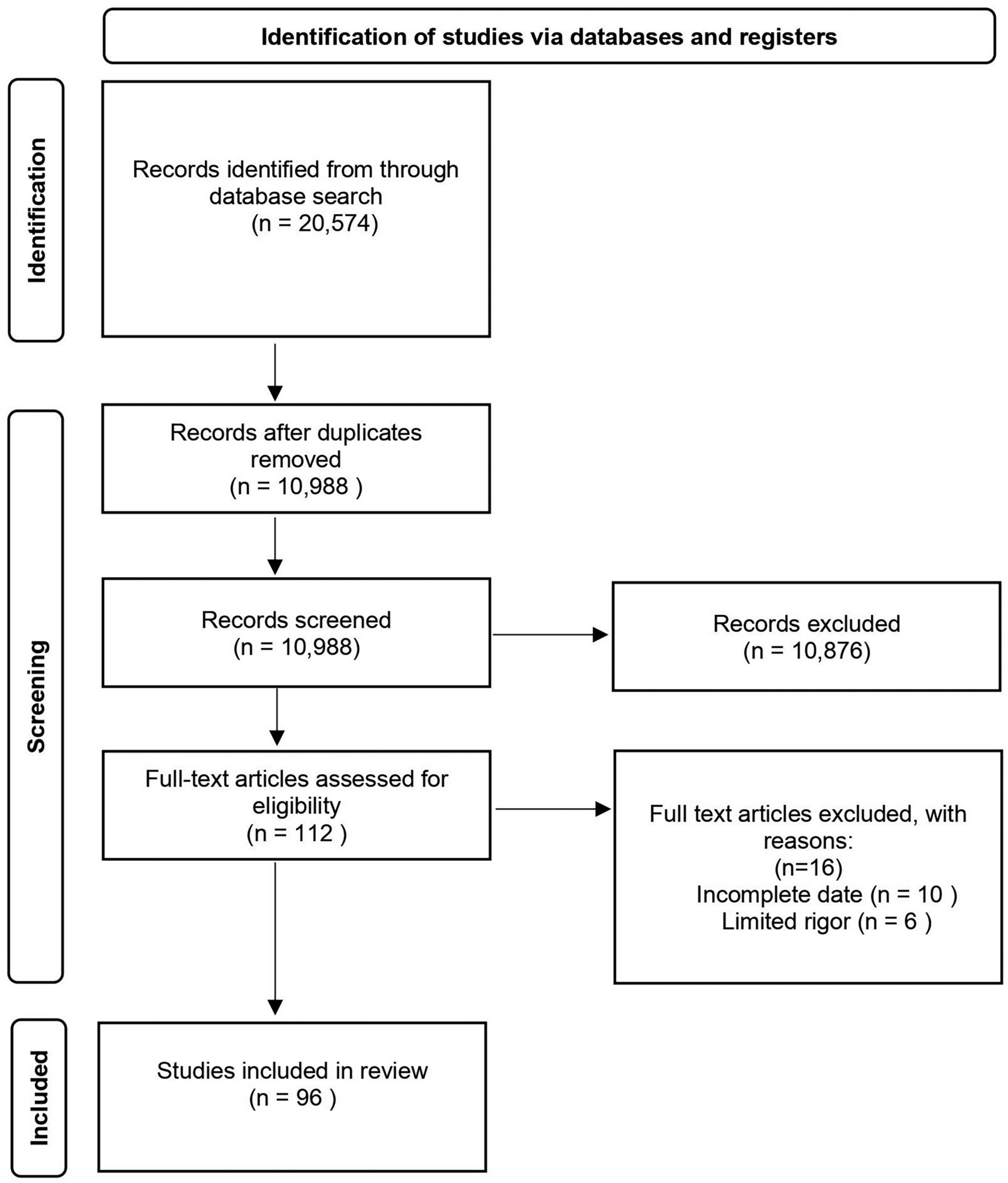
Figure 1 shows the trial flow chart for the patient screening, treatment allocation, intervention, outcomes assessment and data analysis. Figure 2 provides an overview of the study conduct, review, description and interpretation.
Fig. 1
The trial flow chart. TEAS transcutaneous electrical acupuncture stimulation, NRS numerical rating scale, CPSP chronic postsurgical pain, PONV postoperative nausea and vomiting, NE norepinephrine, Cor cortisol, TNF-α tumor necrosis factor, IL-6 interleukin 6
Fig. 2
The schedule of trial enrollment, interventions, and assessments. TEAS transcutaneous electrical acupuncture stimulation, NRS numerical rating scale, CPSP chronic postsurgical pain, PONV postoperative nausea and vomiting, NE norepinephrine, Cor cortisol, TNF-α tumor necrosis factor, IL-6 interleukin 6, POD postoperative day
InterventionsThe TEAS group will receive bilateral Hegu (LI4), Wai guan (TE5), Quchi (LI11), Neiguan (PC6), Chize (LU5), and Taichong (LR3) for electrical stimulation treatment (Fig. 3). LI4 belongs to the Hand Yangming Large Intestine Meridian, which is located at the midpoint of the radius of the second metacarpal bone on the back of the hand. TE5 belongs to the Hand Shaoyang Triple Jiao Classic, which is located in the posterior area of the forearm, 2 in. above the transverse line of the distal dorsal wrist, at the midpoint of the space between the ulna and radius. LI11 belongs to the Hand Yangming Large Intestine Meridian, which is located in the elbow area, at the midpoint depression of the line connecting the ulna and the external epicondyle of the humerus. PC6 belongs to the Hand Jueyin Pericardium Meridian, which is located in the anterior area of the forearm, 2 in. above the transverse line of the distal carpometacarpal side, between the palmaris longus tendon and the radial flexor carpi tendon. LU5 belongs to the Hand Taiyin Lung Meridian, which is located in the elbow area, on the transverse stripe of the elbow, in the concave radial margin of the biceps brachii tendon. LR3 belongs to the Foot Jueyin Liver Meridian, which is located on the dorsum of the foot, between the first and second metatarsals, in the anterior depression of the metatarsal junction. The electrode is attached to the skin surface of the patient’s acupoints and connected to the Huatuo Treatment Instrument (Suzhou Electronic Needle Therapy Instrument SDZ-II) through a wire. TEAS stimulation uses a dense-disperse frequency of 2/100 Hz alternating every 3 s. The waveform is a symmetric biphasic curve. The stimulation intensity will be set to the maximum tolerance level of each patient. TEAS or no TEAS will be performed 30 min before anesthesia induction and each of 7 days postoperatively, with each session lasting 30 min. In the control group, electrodes will be attached to the patients at the same acupoints, but the electrodes will be only connected to the device without electrical stimulation.
Fig. 3
Acupoints selected in this trial: A Hegu (LI4), Waiguan (TE5) and Quchi (LI11); B Neiguan (PC6) and Chize (LU5); C Taichong (LR3); D the Huatuo Treatment Instrument (Suzhou Electronic Needle Therapy Instrument SDZ-II)
All subjects will use the same anesthesia regimen. Anesthesia induction: midazolam 0.05 mg/kg, sufentanil 0.5 μg/kg, etomidate 0.2–0.6 mg/kg, rocuronium 0.6–0.9 mg/kg. Total intravenous anesthesia will be used for anesthesia maintenance: dexmedetomidine hydrochloride 0.4 μg/kg/h, remifentanil 0.05–2 μg/kg/min, propofol 4–12 mg/kg/h, additional rocuronium as needed during surgery, and the bispectral index (BIS) maintained at 40–60. Postoperative patient-controlled intravenous analgesia (PCIA): 30 min before the end of surgery, the PCIA will be connected and the analgesic formula 2 μg/kg sufentanil plus saline to a total volume of 150 ml, 3 ml/bolus, 15-min interval.
We will collect venous blood (5 ml) from patients before the first TEAS treatment and at the end of the operation, and then centrifuge and freeze the plasma supernatant for later testing.
Patients’ vital signs will be constantly monitored during the study, and adverse events will be recorded if they encounter serious adverse reactions and will be promptly discontinued and given symptomatic treatment.
Outcome AssessmentPrimary OutcomeThe primary outcome of this study will be the incidence of CPSP at 3 months after surgery. At 3 months after surgery, the patients will be followed up using a telephonic questionnaire [14, 15]. The questionnaire is provided in Supplementary Material.
Secondary OutcomesThe secondary outcomes will include the incidence of CPSP at 6 months after surgery, the NRS scores at 3 and 6 months after surgery, as well as the NRS scores at 24, 48, and 72 h after surgery. The NRS scores at 3 and 6 months after surgery will be assessed by the outcome evaluator using a telephone questionnaire (see Supplementary Material 1). The NRS scores at 24, 48, and 72 h after surgery will be evaluated by the outcome evaluator at the ward. To observe changes in serum stress factors, such as norepinephrine (NE), cortisol (Cor), and serum inflammatory factors, such as tumor necrosis factor (TNF- α) and interleukin 6 (IL-6). Secondary outcomes will also include remifentanil consumption during general anesthesia, the proportion of rescue analgesic demand at 24, 48, and 72 h after surgery, the duration of thoracic tube indwelling, the incidence of postoperative nausea and vomiting (PONV), and the number of thoracic tubes indwelling at 24, 48, and 72 h after surgery.
Statistical MethodsWe will perform all the analyses in an intention-to-treat population, which includes all patients who have undergone randomization and interventions. Descriptive statistics will be applied to present the characteristics of each group with mean ± standard deviation, median (interquartile), or the number of cases (%). Either analysis of variance or Chi-square test will be used to reveal the baseline balance between groups, depending on the characteristics of the variables. Repeated measures analysis of variance will be applied for repeated data, considering normality and homogeneity. A P value < 0.05 will be considered statistically significant. For missing data, analysis will be performed according to a worst-case scenario. In particular, patients lost to follow-up in the TEAS groups and the sham group will be considered with and without pain, respectively. All data will be analyzed using the statistical package for IBM SPSS Statistics V25.0.
Data Collection Methods and Study MonitoringData collection and management procedures will be developed by statisticians and principal investigators, and the database will be established. The data and safety monitoring committee (DMC) includes a expertise clinical doctor, a scientific research manager and a statistician. All the adverse events should be submitted to the DMC every 6 months. DMC has the right to decide whether to stop or modify the scheme. The scientific research management committee and the statistician will have access to all the data in the study.
Informed ConsentThe researchers will obtain written informed consent from all participants recruited to the study before randomization.
ConfidentialityDuring the study, in order to ensure the privacy and safety of patients, we will use numerical numbers instead of subject names. The personal information of all subjects will be kept strictly confidential and will not be disclosed to any person or organization without authorization.
留言 (0)