Study population and ethical considerations
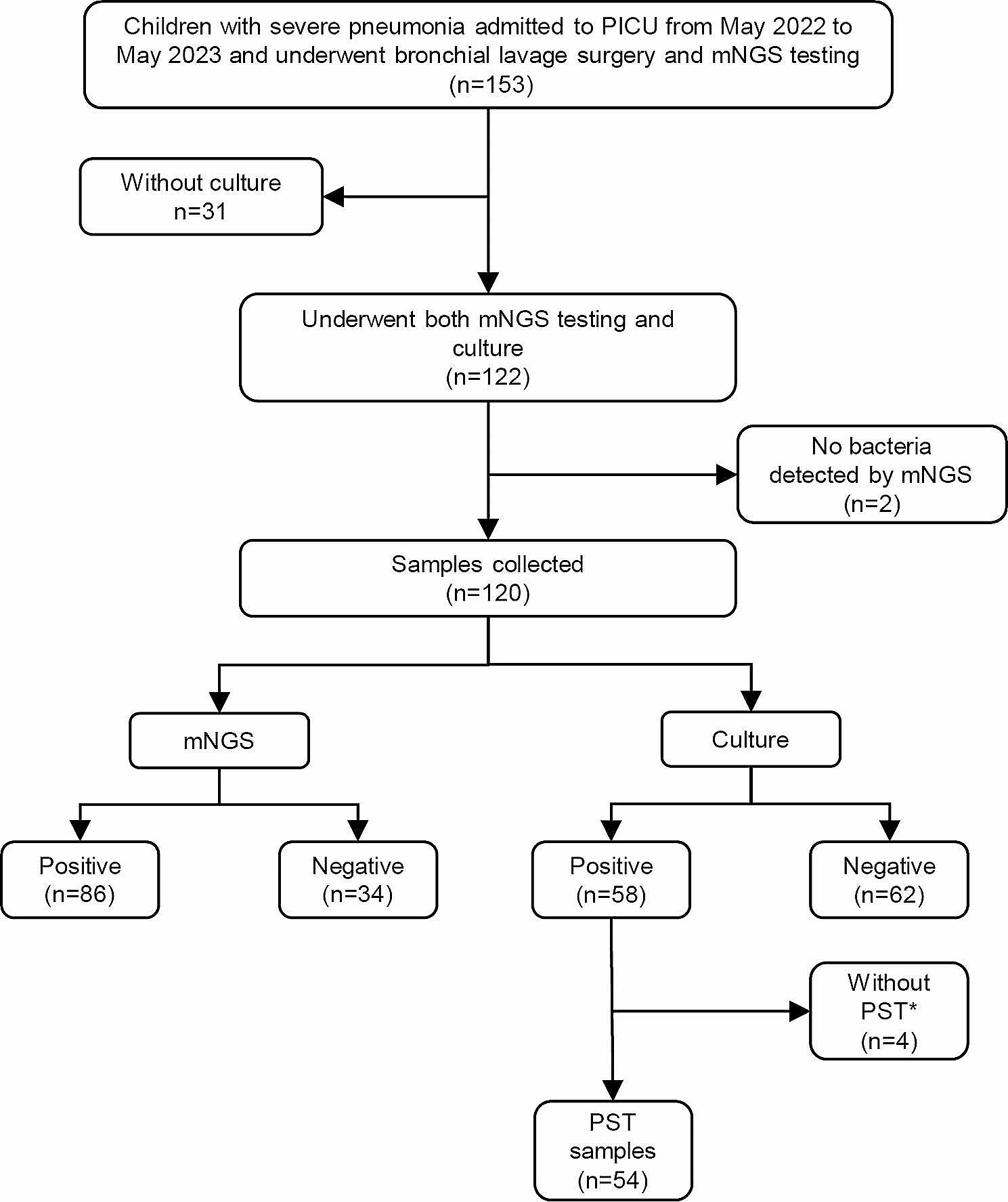
Between June 1, 2020, and November 1, 2022, a total of 583 patients with suspected pulmonary infection were enrolled in this study, originating from four medical institutions located in China. All enrolled patients were required to meet the following criteria: Firstly, they had to present with symptoms such as fever, cough, expectoration, shortness of breath, dyspnea, and abnormal imaging findings. Secondly, bronchoalveolar lavage (BALF) samples were collected concurrently for both metagenomic next-generation sequencing (mNGS) and culture to identify pathogens. Thirdly, the quality inspection and BALF sample testing process met the standards of mNGS. Patients with confirmed Gram-positive bacterial infections, fungal infections, or infections caused by other pathogens were excluded from the analysis. Demographic and baseline characteristics, clinical presentation, radiography and laboratory findings, treatment, and outcomes of the 583 patients were investigated for clinical diagnosis. The study was approved by the Institutional Review Board of the First Affiliated Hospital of Nanjing Medical University (approval no. 2022-SR-014).
Clinical groups of patients
Patients were stratified into three groups based on the presence of co-existing diseases. The Simple Pulmonary Infection Group: This group included patients without underlying diseases. The Immunosuppressed Group: This group comprised patients diagnosed with autoimmune diseases, post-splenectomy individuals, and/or those on long-term treatment with glucocorticoids, immunosuppressive agents, cytotoxic drugs, hematological malignancies, or those who had undergone chemotherapy in the last 6 months or solid organ transplantation. The Chronic Airway Disease Group: This group included patients with chronic bronchitis, bronchiectasis, or chronic obstructive pulmonary disease (COPD), but without immunosuppression.
Specimen collection and processing
Bronchoalveolar lavage fluid (BALF) for metagenomic next-generation sequencing (mNGS) and traditional culture was collected by an experienced clinician using standard procedures from patients with suspected pulmonary infection. Before the procedure, patients’ nasal or oral cavities were cleansed with normal saline. Dexmedetomidine was administered for sedation before the bronchoscopy, and local anesthesia with 2% lidocaine was applied during the examination. The electronic bronchoscope was used to examine all bronchi in detail, and any lesions found on a chest CT scan were brush-examined before BALF collection. The area for lavage was determined based on the lesion area found on the chest CT scan, with BALF collected from the right middle lobe or the subsegment of the left lingual lobe if scattered lesions were present.
Approximately 100 mL of sterile normal saline was injected into the target bronchus in batches at 37 °C, with the first 20 mL discarded to avoid contamination, and approximately 5 mL collected into sterile tubes. The BALF samples were then divided into aliquots for pathogen detection, with one aliquot inactivated (56 °C, 30 min) before nucleic acid extraction.
mNGS assay
(i) Nucleic Acid Extraction: Bronchoalveolar lavage fluid (BALF) samples were procured following standard protocols. DNA extraction employed the TIANamp Micro DNA Kit (Tiangen Biotech, Beijing, China), adhering to the manufacturer’s guidelines. Each batch included a no-template control (NTC) alongside clinical specimens. DNA quantification and quality assessment utilized Qubit and NanoDrop devices (Thermo Fisher Scientific). (ii) Library Preparation and Sequencing: The Hieff NGS C130P2 OnePot II DNA Library Prep Kit for MGI (Yeasen Biotechnology) facilitated DNA library construction, in line with manufacturer instructions. Post-preparation, libraries underwent Agilent 2100 qualification and were sequenced as 50 bp single-ends on DNBSEQ-200 (MGI Tech, China). Quality control (QC) criteria required over 18 ng of DNA post-library construction and a minimum of eight million raw reads. (iii) Bioinformatics Analysis: An in-house bioinformatics pipeline was employed for microorganism identification. Quality sequencing data were refined by removing low-quality reads, adapter contaminants, duplicates, and reads shorter than 36 bp. Human sequences were filtered out using bowtie2 software against the hs37d5 human reference genome. The residual data was aligned with the NCBI microorganism genome database using Kraken2, enabling the determination of the samples’ microbial composition.
Culture method
The BLAF was inoculated onto bacteriological media, including blood agar, chocolate agar, and blue agar plates, using sterile wire loops. Incubation was carried out at 35 °C for 48 h in a 5% CO2 atmosphere within a thermostatic incubator. Dominant colonies were then selected for bacterial identification using the VITEK2-Compact, an automated system from BioMerieux, France. Bacterial strains identified in the BALF at concentrations of ≥ 10^3 colony-forming units per milliliter were deemed causative pathogens.
Three interpretational approaches of mNGS
Simple Interpretation (SI): Gram-negative bacteria were identified by mNGS. Laboratory Interpretation (LI) [18, 19]: the parameter of gram-negative bacteria reached one of the following criteria: (i) relative abundance of pathogens detected by mNGS at the genus level was greater than or equal to 30%, regardless of the culture results, or (ii) the coverage rate scored 10-fold greater than that of any other microbes according to Langelier’s study. Laboratory interpretation serves as the positive standard for mNGS. Clinical Interpretation (CI): the bacteria identified in LI were further screened based on additional criteria. This microbe must have unambiguous literature evidence of its pulmonary pathogenicity, and the matched patient must have had risk factors for its infection.
Clinical diagnosis
Two physicians with expertise in managing infections (WKS and XSC) conducted an independent review of all patient medical records, as well as the results of culture and mNGS. The physicians initially determined whether patients had an infectious or noninfectious etiology. Following this, they identified the causative pathogens by evaluating a combination of clinical manifestations, laboratory tests, chest radiology, and microbiological tests (including culture and mNGS). Any disagreements between the two intensivists were resolved through in-depth discussion, and another senior physician (SLD) was consulted if consensus could not be reached.
Statistical analysis
Following the extracted data, 2 × 2 contingency tables were derived to determine sensitivity, and the McNemar test was used for discrete variables when appropriate. Differences between qualitative variables were assessed using the Fisher exact test, and the chi-square test was used to compare differences in positivity rates. Concordance was assessed using kappa statistics (kappa ≤ 0.4 low, kappa 0.41–0.6 fair, kappa > 0.6 good). Data analyses were performed using SPSS18 and GraphPad Prism7 software. P values smaller than 0.05 were considered statistically significant, and all tests were two-tailed.

留言 (0)