
記住我
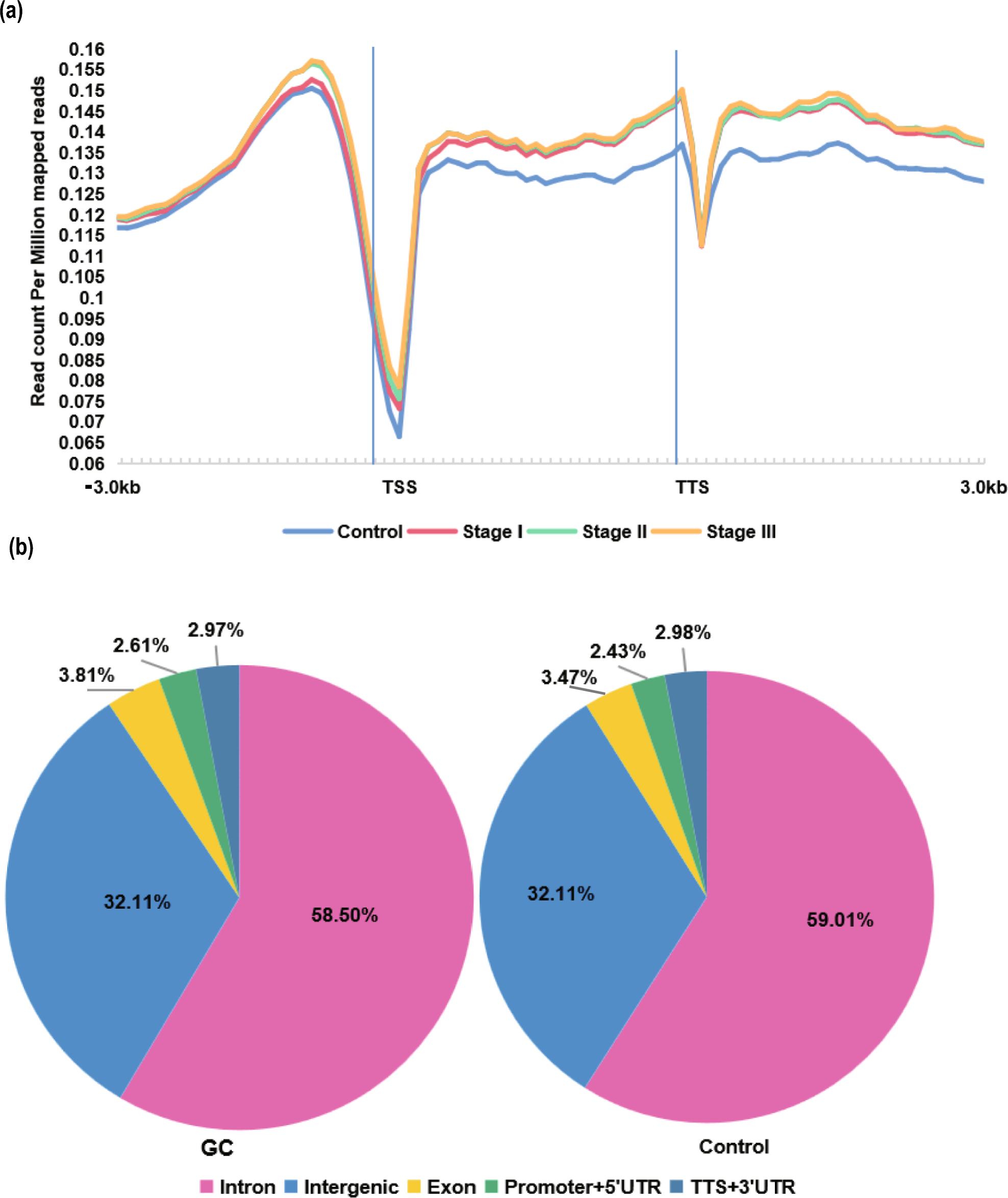


To capture the biological and biochemical characteristics of the different KRAS mutants in our GC PDX platform, we first evaluated their distribution among the 200 profiled available models. KRAS mutations have been detected in 23 PDXs (12% of PDXs); among them, the most frequent mutations were G13D (11, 47.8%), G12D (7, 30.4%), A146T (3, 13%), G12C (1, 4.3%) and G12V (1, 4.3%) (Fig. 1a). The distribution of KRAS variants was found in line with the percentage of mutants reported in the TCGA cohort (7.3% KRAS-mutated patients of which 47.6% G13D, 33.3% G12D, 9.5% A146T, 4.7% G12C, 4.7% G12V mutants), underlining the potential of this PDXs GC collection to capture KRAS mutational status in this malignancy (Fig. 1b). Interestingly, as previously reported [26], the frequency of KRAS mutations is increased in PDXs due to higher engraftment rate but the ratio of the different mutants is not altered.
Fig. 1
The GC platform is representative of the KRAS mutants in the TCGA cohort. Pie charts showing the percentage of KRAS-mutated patient-derived xenografts (PDX) present in the PDX Gastric Cancer platform (a) and in the TCGA cohort (b)
GC KRAS-mutated cells display different KRAS-GTP levels but similar addiction to the KRAS gene.It is known that gain-of-function missense mutations increase KRAS GTP levels [28]. Since the level of activation of the KRAS A146T is largely unknown, we performed G-LISA RAS activation assay (Fig. 2a) on PDX-derived primary cells (3 KRAS A146T, 2 KRAS G12D and 1 KRAS G13D models) (Suppl. Figure 1a). Since the availability of A146T gastric primary cellular models was limited, we included two colorectal cancer cell lines bearing this less frequent mutation (SNU81 mutated in a single allele and LS1034 lacking the WT allele). As shown in Fig. 2a, we observed that in GTR0245 cells, presenting a homozygous G12D mutation (Suppl. Figure 1b), the level of active KRAS was higher than in GTR0249 cells, carrying the same mutation in heterozygosis. Similarly, the presence of the A146T mutation in homozygosis (in LS1034 cells) resulted in a level of RAS activation higher than in the heterozygous counterpart. This result suggests that the co-existence of two mutated alleles leads to a more potent KRAS activity. Interestingly, the KRAS activation state in A146T mutated models, both in heterozygosis and homozygosis, was lower than that of cells mutated for G12/13D.
Fig. 2
KRAS mutants display different levels of activation but similar addiction to the KRAS oncogene. a Bar graph comparing RAS-GTP levels in KRAS-mutated models. Values in fold change of RAS-GTP levels are relative to 293T cells (WT for KRAS). Filled bars indicate homozygous mutants; dashed bars represent heterozygous mutants. b Cell viability upon KRAS silencing was evaluated in five KRAS-mutated gastric cancer models using crystal violet staining. Both histograms show mean ∓ SD of at least three independent experiments. sh short hairpin; (Student’s t test) *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001
In spite of the different KRAS activation status, however, cells were equally addicted to the different mutants, as shown by in vitro silencing experiments, in which we transduced five mutated models with two different KRAS short hairpin RNAs (shRNAs) (Fig. 2b and Suppl. Fig b). In sum, our results show that, regardless the type of mutation and zygosity, KRAS-mutated cells significantly rely on the activity of this oncogene for survival, suggesting that its inhibition might lead to a therapeutic response in all the mutated cases.
KRAS-mutated models response to KRAS downstream inhibitors depends on their intrinsic molecular landscapeOver the years, many research groups have put their efforts in tackling this ‘difficult-to-target’ oncoprotein, implementing direct and indirect strategies to target KRAS mutants. To study whether KRAS G12D and A146T mutants were able to differentially activate KRAS downstream pathways, we explored their sensitivity to the MEK-inhibitor (MEKi) Trametinib, alone or in combination with the AKT inhibitor MK-2206.
As shown in Fig. 3, while models harbouring the G12D mutation (GTR0245 and GTR0249) benefitted of the drug combination, a strong effect of Trametinib alone was observed in two of the A146T mutated models (GTR0213-heterozygous- and LS1034-homozygous-), suggesting a vulnerability for this mutant at low doses of the MEKi. On the other hand, in the GTR0128 and SNU81 models (A146T heterozygous mutants) the Trametinib/MK-2206 combination resulted in a remarkably increased response, likely due to the additional presence of point mutations in PIK3CA (GTR0128) and PTEN (SNU81) genes, respectively (Table 1). Interestingly, the interrogation of gastric cancer databases showed that the co-occurrence of KRAS and PIK3CA pathway mutations is around twofold more frequent for A146T than for the other mutants (66% vs 23%, Suppl. Figure 4).
Fig. 3
KRAS A146T models show a strong sensitivity to Trametinib in vitro treatment, in the absence of additional driver mutations. Heatmap showing the viability of different KRAS mutant PDXs treated for 72 hours with the indicated doses of the single agents Trametinib or MK-2206 or the combo. The average of three independent experiments is shown. The scale represents the percentage of viable cells
Table 1 Summary of relevant mutations detected in KRAS A146T modelsTo confirm our in vitro results and validate the sensitivity of A146T-mutants to Trametinib in the absence of concomitant driver mutations, we performed preclinical trials on the PDXs from which the primary cell lines have been generated (Fig. 4). Briefly, KRAS-mutated PDXs were passaged until production of a cohort of 40 mice. Established tumours (average volume, 300 mm3) were randomized and treated with Vehicle, Trametinib, MK-2206, either as single agents or in combination. Results confirmed the sensitivity of GTR0213 tumours (A146T-mutated model) to Trametinib alone, showing no statistical difference between the single arm of MEKi and the combination treatment (Fig. 4a). In agreement with in vitro experiments, the combo was the only effective treatment in the context of the GTR0128 PDX, in which the A146T mutation is concomitant with a PIK3CA mutation (Fig. 4b). Likewise, the GTR0245 model (G12D homozygous) showed a statistically significant difference between the Trametinib monotherapy and the combination (Fig. 4c).
Fig. 4
In vivo effectiveness of the single agent Trametinib in KRAS A146T mutants without co-occurent PIK3CA/PTEN mutations. a–c Tumor growth curves of mice cohorts derived from GTR0213, GTR0128 and GTR0245 PDXs, treated with the MEKi inhibitor Trametinib and the AKT inhibitor MK-2206, alone or in combination, as indicated. The different inhibitors were used at the following doses: Trametinib 1 mg/kg, daily, per os; MK-2206 100 mg/kg, 3 times per week, per os. Lines represent average tumor volumes + SD for at least 5 animals. Statistical significance was calculated using the Two-way ANOVA with Bonferroni correction. ns not significant, **p < 0.01; ***p < 0.001; the black arrows indicate the start of the treatment. d KRAS and co-occurent mutations for each preclinical model
Biochemical (Fig. 5a) and immunohistochemical (Fig. 5b) analyses confirmed the high sensitivity of two A146T mutated models (GTR0213-heterozygous- and LS1034-homozygous-) to Trametinib monotherapy, showing its ability to abrogate the activation of the PI3K/MAPK downstream effector S6 kinase. On the contrary, G12D-mutated models needed the combo treatment to show the same effect on PS6.
Fig. 5
In vitro and in vivo signal transduction properties of KRAS mutants upon treatment with Trametinib, MK-2206 or the combo. a Western blot analysis of KRAS-mutated models upon 6 h-treatments with Trametinib, MK-2206 or the combo. Vinculin probing was used as loading control b PS6 immunohistochemistry staining of tumor slices obtained from mice receiving vehicle or acute treatments (2 days) with Trametinib, MK-2206 or combo. Magnification: ×40
This striking effect of Trametinib on PS6 in GTR0213 and LS1034 A146T-mutated models, may be due to the previously reported [5, 29] “weakness “of this allele to induce KRAS downstream signals. Conversely, A146T-mutated models (such as GTR0128 and SNU81) displaying additional and “strong” driver mutations (PI3KCA and PTEN respectively) needed the combo treatment to downregulate PS6 (Fig. 5a, b).
Inhibition of the guanine exchange factor SOS1 does not significantly affect the viability of KRAS A146T mutantsAs already described by Poulin et al. [5], KRAS A146T mutants are characterized by a protein structure that does not impair the activation of the GTPase, thus promoting a high rate of intrinsic and GEF-induced nucleotide exchange. We thus investigated if the abrogation of the GEF SOS1 could differentially affect the viability of KRAS A146T mutants compared to the canonical G12D mutants. Unexpectedly, silencing experiments (Suppl. Figure 3) showed that the viability of the A146T mutated models was not significantly affected by SOS1 silencing, regardless of their zygosity. The same result was obtained for the GTR0245 model, homozygous for the G12D mutation, while the GTR0249 (carrying the same mutation in heterozygosis) showed a modest reduction of cell proliferation upon SOS1 silencing (Fig. 6), in agreement with what shown by Wong et al. [25]. Thus, our results demonstrate that, even if the GEF SOS1 is considered a key player in the activation of KRAS A146T mutants, its abrogation does not significantly affect their viability, suggesting that pharmacological targeting of this protein is unlikely to be effective.
Fig. 6
SOS1 silencing does not significantly affect viability of KRAS A146T mutants. Bar graph representing the percentage of cell viability of KRAS-mutated models 48 h upon transfection with SOS1 siRNA. Cell viability was measured using Cell Titer Glo cell viability assay. Bar graphs display mean ± SD; comparisons were made using Student’s t test; ns not significant; ****p < 0.0001
留言 (0)