
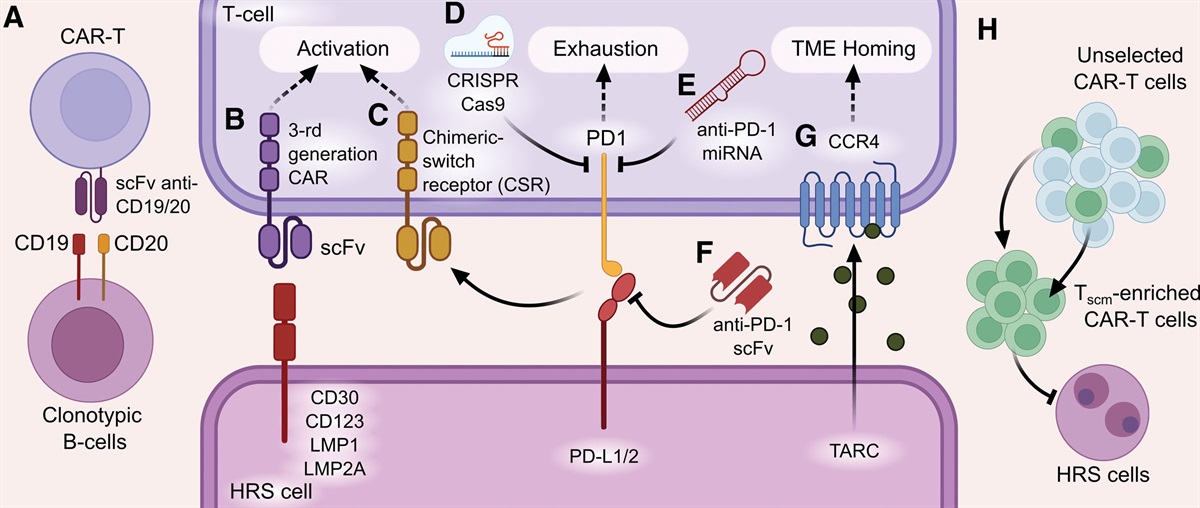
記住我



Aplastic anemia (AA) is a rare and life-threatening disease characterized by pancytopenia and hypoplastic bone marrow (BM).1 Unlike the inherited AA characterized by BM failure in association with one or more somatic abnormalities, most cases of acquired AA are without any identifiable causes (idiopathic). Although the exact pathogenesis of AA is not completely understood, immune-mediated destruction of hematopoietic stem cells (HSCs) following systemic immune dysregulation has emerged as a main mechanism.2 The most relevant evidence for an immune-mediated mechanism is improvement in blood counts following immunosuppressive therapies (IST).1 The treatment for patients with severe AA (SAA), who are ineligible for hematopoietic stem cell transplantation, aims to suppress the cytotoxic T-lymphocyte response with IST using a combination of antithymocyte globulin (ATG) and cyclosporine.3 However, typical response rates to IST are approximately 60%–70%, and one-third of responders eventually relapse,4 suggesting complex cellular factors linked to AA pathogenesis and the IST resistance mechanism.5
As AA patients have diverse genetic, pathogenic, and immunological backgrounds, it is imperative that the comprehensive characterization of cellular factors for IST response is researched thoroughly. Recently, single-cell RNA sequencing (scRNA-seq) has been widely appreciated in relation to its ability to measure rare or heterogeneous cell populations.6 Lundgren et al highlighted the reduction of cytotoxic STAT3-mutated CD8+ T cells by IST.7 Zhu et al reported the inactivation of cellular interactions mediating the destruction of hematopoietic stem and progenitor cells (HSPCs) by cytotoxic T cells after IST.8 The contribution of changes in B cell gene expression and receptor diversity to AA pathogenesis was also indicated by Tonglin et al.9 However, these studies were limited by the number of either cell types or samples, and a comprehensive understanding of AA pathogenesis and the IST response mechanism is still lacking. Hence, this study analyzed cellular diversity and interactions in the BM of healthy controls and AA patients using scRNA-seq. We identify multiple cellular factors associated with AA pathogenesis and IST resistance.
MATERIALS AND METHODS PatientsWe initially collected consecutive BM samples and clinical data from 18 patients with AA and 10 healthy controls between October 2017 and January 2020 from Seoul St. Mary’s Hospital, Seoul, Republic of Korea (Suppl. Table S1). The samples of 2 of the subjects who were diagnosed with nonsevere AA were excluded from scRNA-seq analysis. The quality control indicated that the BM samples of 5 patients with SAA and 7 healthy controls were inadequate for scRNA-seq analysis. The final cohort included 11 SAA patients and 3 healthy controls and was subjected to scRNA-seq. The diagnosis and severity of idiopathic AA were assessed according to previous reports.10,11 Patients received IST consisting of rabbit ATG (Thymoglobulin; Genzyme-Sanofi, Lyon, France) and cyclosporine (n = 7) as previously described12,13 or cyclosporine monotherapy (n = 4). Patients with SAA received the combination of ATG and cyclosporine as the standard treatment. However, some patients with older age (≥65 y) and/or severe comorbidity related to SAA were treated with cyclosporine monotherapy based on the physician’s discretion. A complete response was defined as hemoglobin ≥10 g/dL, absolute neutrophil count ≥1.0 × 109/L, and platelet count ≥100 × 109/L for >8 weeks without transfusion support. A partial response was determined as no longer meeting the criteria for SAA and having transfusion independence for >8 weeks.3,14 An analysis of glycosylphosphatidylinositol-deficient granulocytes was performed by flow cytometry outlined in our previous report.13 Bone marrow samples of healthy controls were collected from healthy donor for allogeneic BM transplantation following written consent. These were obtained during bone marrow harvesting. The protocol was approved by the institutional review board committees of the Seoul St. Mary’s Hospital (IRB number: KC17TESI0426 and KC23SASI0369). The study was conducted in accordance with the Declaration of Helsinki. The scRNA-seq experiment and flow cytometry analysis were carried out using frozen BM, taken from either AA patients at diagnosis before IST or healthy controls.
Single-cell library preparation and sequencingscRNA-seq libraries were prepared using the Chromium Single-Cell 3′ Reagent Kits (v2) comprising a Single-Cell 3′ Library & Gel Bead Kit v2 (PN-120237), Single-Cell 3′ Chip Kit v2 (PN-120236), and an i7 Multiplex Kit (PN-120262) (10× Genomics, Pleasanton, CA, USA) and following the Single-Cell 3′ Reagent Kits (v2) User Guide (manual part no. CG00052 Rev A). The libraries were run on a HiSeq X Ten System (Illumina, San Diego, CA, USA) as 150-bp paired-end reads with 1 sample per lane. All the remaining procedures, including library construction, were performed according to the manufacturer’s protocol.
Antibodies and flow cytometryAll antibodies were against human antigens. Anti-TFRC-Alexa Fluor 700 (M-A712), anti-CD4-APC-cy7 (RPA-T4), anti-Prom1-FITC (567029), and anti-CD34 were purchased from BD Biosciences (Franklin Lakes, NJ, USA). Anti-CD3-Brilliant Violet421 (OKT3) and anti-CD8a-APC (300912) were purchased from BioLegend (San Diego, CA, USA). For the phenotypic analysis of BM cells, 1 × 105 cells were suspended in 100 μL Hanks’ balanced salt solution (Mediatech) containing 2% fetal bovine serum. For each sample, cells were incubated with 1 μg of each fluorescent-conjugated primary antibody for 15 minutes at room temperature and washed with Hanks’ balanced salt solution/fetal bovine serum. After being washed with chilled binding buffer, the cells were incubated with streptavidin-PE on ice for 10 minutes before they were washed and analyzed. Staining data were collected using a FACSCantoTM II Cytometer (BD Biosciences).
Clustering analysis and gene set enrichment testFurther scRNA-seq analysis was performed using R (version 4.0.2). We used the FindVariableFeatures in the Seurat package (version 3.2.0) to identify highly variable genes and then performed principal component analysis with the top 2000 variable genes based on the default method, “vst.” Clusters were partitioned with FindClusters in the Seurat package, and cells were projected into a two-dimensional space with uniform manifold approximation and projection based on the default method, “matrix.” The parameters used for clustering analysis are listed in Suppl. Table S2. The differentially expressed genes (DEGs) in each cluster were identified using FindMarkers in the Seurat package based on the default test, “wilcox.” All processes were performed with default options in the Seurat package. We also used the SingleR15 method, which is based on the correlation of gene expression between single cells and a homogenous cell population, with the Novershtern Hematopoietic Data database providing 211 human microarray samples annotated to 16 main types and 38 subtypes of cells. To remove batch effect when integrating the scRNA-seq from the other database with our data, we used the RunHarmony in the Harmony package16 (version 0.1.1) with default parameter while considering the origin of data as batch information, Gene set enrichment test was performed with the Database for Annotation, Visualization, and Integrated Discovery website.17,18 The top 5 gene ontology terms, ranked by P values, are described in our results.
Trajectory analysisWe performed single-cell trajectory analysis using the Monocle2 package (version 2.18.0).19 To chronologically sort cells by pseudotime using the orderCells in Monocle2, we selected the top 2000 DEGs identified by the FindAllMarkers in the Seurat package, based on the default test, “wilcox.” To visualize and interpret the results using the plot_cell_trajectory in Monocle2, the dimension was reduced using reduceDimension in Monocle2 based on the DDRTree method. The other parameters were provided with default options.
Cellular interaction analysisWe performed cell–cell interaction analysis using CellPhoneDB,20 which is a public repository of interactions between ligands and receptors. We used the python (version 3.7.0) and cellphonedb method in CellphoneDB python package (2.1.2) based on the default parameters for the analysis, and the count per million normalized single-cell expression data of HSCs, T cells, and B cells from all samples were used as the input. Interactions of receptors and ligands between 2 cell types were predicted based on the expression profiles of receptors of 1 cell type and the expression profiles of the corresponding ligand of another cell type.
Statistical analysisAll statistical tests were performed on directed pairwise comparisons. Unpaired t-tests were performed to analyze differences between 2 groups. No mathematical corrections were made for multiple comparisons. Significance is displayed as *P < 0.05; **P < 0.01; ***P < 0.001.
RESULTS Patient characteristicsThe median age of the patients was 52 years (range 22–67). Out of total, 8 were classified as SAA, whereas the severity of 3 AA patients was very SAA. Seven patients received IST with ATG and cyclosporine, and 4 patients received cyclosporine monotherapy. Five IST responders (R1–4, 7) achieved a complete response at 8.9, 6.1, 2.3, 11.6, and 24.4 months, respectively. Two IST responders (R5–6) achieved a partial response at 14.3 and 4.3 months, respectively. Four patients had no response (N1–4). Eight patients had small paroxysmal nocturnal hemoglobinuria clones in glycosylphosphatidylinositol-deficient granulocytes (R2–4, R6–7, N2–4). All patients did not have a family history of BM failure syndrome as well as pregnancy/virus-related onset. In 2 patients with onset age younger than 40 years (R2 and R3), there was no evidence of Fanconi anemia by assessment of chromosome breakage on peripheral blood lymphocytes. All patients were classified as having idiopathic AA with normal cytogenetics. Next-generation sequencing-based gene panel testing using peripheral blood did not reveal any pathogenic variants representing inherited BM failure syndrome (Suppl. Tables S3 and S4).21,22
scRNA-seq of BMs from healthy controls, IST responders, and IST non-responders characterizes differential proportion of diverse cell typesTo depict the baseline cellular composition, lineage, and interaction of BM cells from patients with AA and their relation to IST responses, we profiled BM cells from 11 treatment-naive patients with AA, including 7 IST responders (R1–R7), 4 non-responders (N1–N4), and 3 healthy controls (H1–H3) using droplet-based scRNA-seq (Figure 1A). After a quality control, a total of 67,271 cells were retained, comprising 33,646 cells from IST responders (mean: 4807 cells per patient), 21,166 cells from non-responders (mean: 5292 cells per patient), and 12,459 cells from healthy controls (mean: 4153 cells per patient) (Suppl. Table S5). Subsequently, we performed unsupervised clustering analysis and used uniform manifold approximation and projection (UMAP)23 to visualize the cells according to cell type (Figure 1B) and IST response (Figure 1C). We classified our clusters into 8 broad cellular lineages: HSCs, megakaryocytes (MKCs), myeloid cells, myeloid natural killer (NK) cells, plasmacytoid dendritic cells (pDCs), erythroid precursor cells (EPCs), B cells, CD4+ T cells, CD8+ T cells, and lymphoid NK cells by combining the results of the SingleR package in R and the expression profiles of cell type-specific marker genes (Figure 1D). Notably, HSCs and pDCs were significantly depleted in both IST responders and non-responders compared with healthy BM (Figure 1E,F; Suppl. Figure S1; Suppl. Table S6). EPCs were marginally depleted in IST non-responders compared with healthy controls (P = 0.058; Figure 1E,F). We found an increase of CD8+ T cells in IST responders compared with healthy controls (P = 0.1; Figure 1E,F).
 Figure 1.:
Figure 1.: scRNA-seq of healthy controls and patients. (A) Workflow of the study design. scRNA-seq was conducted with bone marrow cells extracted from 3 healthy controls and 11 patients (7 IST responders and 4 non-responders). (B) UMAP plot of clusters including all samples. (C) UMAP plots of the group-specific distribution of cells. (D) Heatmap representing cluster-specific gene expression. The yellow color represents high expression and purple represents low expression. (E) Proportion of clusters (top) and number of cells (bottom) in each sample. (F) Box-and-whisker plot comparing the proportions of clusters among healthy controls, IST responders, and non-responders. The solid horizontal line represents the median, the lower boundary represents the lower quartile, the upper border represents the upper quartile, and the whiskers represent outliers (t test, *P < 0.05). AA = aplastic anemia; EPC = erythroid precursor cell; HSC = hematopoietic stem cell; IST = immunosuppressive therapy; MKC = megakaryocyte; NGS = next generation sequencing; NK = natural killer; pDC = plasmacytoid dendritic cell; scRNA = single-cell RNA; UMAP = uniform manifold approximation and projection.
Depletion of early-stage EPCs was observed in AA patientsAlthough the depletion of erythrocytes in the peripheral blood of AA patients was well known,24 the distribution of EPCs in the BM of AA patients is not fully understood. We performed reclustering analysis of EPCs and identified 10 subclusters that were largely grouped based on erythroid genes, such as CD34, GYPA, and TFRC (Figure 2A). Based on our trajectory analysis, they were divided into early-stage (C1, C9), middle-stage (C0, C2, C3, C6, C7, C8), and late-stage (C4, C5) EPCs (Figure 2B; Suppl. Figure S2). Interestingly, the CD34+GYPAloTFRC+ subcluster (C9) was significantly depleted in AA patients (Figure 2C–F; Suppl. Figures S2 and S3). Using flow cytometry, we confirmed the significant depletion of CD34+TFRC+ EPCs in AA patients (Figure 2G,H).
 Figure 2.:
Figure 2.: EPC subclusters. (A) UMAP plot of the EPC subclusters. (B) Trajectory plots of the group-specific distribution of EPC subclusters. The arrow represents the direction of differentiation. (C) UMAP plots of the group-specific distribution of EPCs. (D) Box-and-whisker plot comparing the proportion of cells within EPC subcluster 9 between healthy controls and patients (t-test, ***P < 0.001). (E) Box-and-whisker plot comparing the proportion of cells within EPC subcluster 9 among healthy controls, IST responders, and IST non-responders (t-test, ***P < 0.001). (F) UMAP plots of the expression of marker genes characterizing EPC subclusters (zoomed-in views represent subcluster 9). (G) Representative images for EPC subclusters using flow cytometry. (H) Box-and-whisker plot comparing the percent of CD34+ EPC among healthy controls, IST responders, and non-responders. The solid horizontal line represents the median, the lower boundary represents the lower quartile, the upper border represents the upper quartile, and the whiskers represent outliers (t-test, ***P < 0.001). AA = aplastic anemia; BM = bone marrow; EPC = erythroid precursor cell; IST = immunosuppresive therapy; UMAP = uniform manifold approximation and projection.
HSCs with low expression of PROM1 were partially depleted in AA patientsAs described above, a significant depletion of HSCs was observed in AA patients. To understand the cellular heterogeneity of HSCs related to this phenomenon, we reclustered HSCs into 6 subclusters, which were largely divided into 2 groups based on PROM1 expression (Figure 3A,B; Suppl. Figure S3).25–27 PROM1+ HSCs (C2, C4, C5) were almost completely depleted in AA patients, but PROM1−/low HSCs (C0, C1, C3) were retained in AA patients (Figure 3B,C). Furthermore, the diversity of HSC subclusters decreased significantly in AA patients compared with healthy controls (Suppl. Figure S4B). Our trajectory analysis showed 2 distinct differentiation pathways for the PROM1+ and PROM1−/low HSC lineages (Figure 3D; Suppl. Figure S5). We analyzed DEGs and found that the partially depleted PROM1−/low HSCs exhibited a relative upregulation of autophagy-related genes implicated in HSC survival (Figure 3B).28 Furthermore, the relative upregulation of inhibitory checkpoints, such as those encoded by BTLA,29,30CYBB,31,32 and TNFRSF14,33,34 and the downregulation of cytokine receptor IL6R35,36 (Figure 3E) in the partially depleted PROM1−/low HSCs represents the ability of immunosurveillance. To confirm the association of PROM1 to the depletion of HSPCs in AA patients from an independent scRNA-seq cohort, we reclustered HSPCs based on data from Zhu et al (Figure 3G).8 One HSPC subcluster (C6) with relatively high expression of PROM1 was identified as depleted in AA patients (Figure 3H,I), which was consistent with our results. For further validation, we checked the expression of PROM1 in CD34+ hematopoietic cells from healthy controls, IST responders, and non-responders using flow cytometry (Figure 3J). Notably, PROM1 expressing CD34+ hematopoietic cells were significantly depleted in both IST responders and non-responders (Figure 3K).
 Figure 3.:
Figure 3.: HSC subclusters. (A) UMAP plot of HSC subclusters. (B) Heatmap of the expression profile of marker genes [z score of log2(count per million+1)] used to identify the cell type of each subcluster and to determine the degree of depletion. Red and blue colors denote upregulated and downregulated genes, respectively. (C) Proportions (top) and cell counts (bottom) of HSC subclusters for each sample (HSCs were not detected in R4, R5, N3, and N4). (D) Trajectory plot depicting the degree of differentiation and cluster distribution of all samples by pseudotime analysis. The arrow represents the direction of differentiation. (E) Violin plots of the expression profile of DEGs representing the characteristic of PROM1−/low HSCs. (t-test, *P < 0.05, ***P < 0.001). (G) UMAP plot of HSPC subclusters, (H) Dot plot of the expression profile of PROM1. (I) Box-and-whisker plot comparing the proportions of clusters between healthy controls and AA patients. The solid horizontal line represents the median, the lower boundary represents the lower quartile, the upper border represents the upper quartile, and the whiskers represent outliers (t-test, **P < 0.01). (J) Representative histograms show the expression of PROM1 of CD34+ HSCs. (K) Box-and-whisker plot comparing the percent of PROM1+ HSCs among healthy controls, IST responders, and non-responders. The solid horizontal line represents the median, the lower boundary represents the lower quartile, the upper border represents the upper quartile, and the whiskers represent outliers (t-test, ***P < 0.001). AA = aplastic anemia; BM = bone marrow; DEG = differentially expressed genes; HSC = hematopoietic stem cell; HSPC = hematopoietic stem and progenitor cells; IST = immunosuppressive therapy; UMAP = uniform manifold approximation and projection.
Enrichment of CD8+ T cells in IST respondersWe further investigated diverse subclusters of T cells. As a result, we identified 13 T cell subclusters: CD4+ T cells (C0, C1, C6, C8, C10), 4 CD8+ T cells (C2, C4, C9, C12), 1 NK cell (C3), 2 Tregs (C5, C7), and 1 CLP (C11) (Figure 4A) according to the expression patterns of major T cell lineage markers (Figure 4B). Additionally, based on the top markers of each subcluster and the results from the trajectory analysis, we further annotated each subcluster with 4 characteristics, such as naive, cytotoxic, exhausted, and memory (Suppl. Tables S7 and S8 and Figure S6). Although the diversity of the T cell subclusters was similar among healthy controls, IST responders, and non-responders (Suppl. Figure S7A), we found that IST responders showed marginally enriched CD8+ T cells compared with healthy controls (P = 0.075; Figures 1F and 4C,D). Further verification through flow cytometry using 18 BMs of IST responders (n=9) and non-responders (n=9) confirmed that IST responders also showed significantly higher CD8+ T cell rates compared with non-responders (Figure 4E,F). We also found the significant enrichment of CD8+ T cells in IST responders by in silico sorting T cells expressing CD8A from scRNA-seq data (Figure 4G).
 Figure 4.:
Figure 4.: T cell subclusters. (A) UMAP plot of T cell subclusters (NK, Treg, and CLP). (B) Heatmap of the expression of marker genes used to identify the cell type of each subcluster. Red and blue colors denote upregulated and downregulated genes, respectively. (C) Proportion of T cell subclusters in each sample. (D) Box-and-whisker plot comparing the proportion of CD8+ T cell subclusters among healthy controls, IST responders, and non-responders. The solid horizontal line represents the median, the lower boundary represents the lower quartile, the upper border represents the upper quartile, and the whiskers represent outliers (t-test). (E) Representative images of CD4+ T cells and CD8+ T cells from the BMs of IST responders and non-responders through flow cytometry analysis. (F) Box-and-whisker plot comparing the percent of CD8+ T cells between IST responders and IST non-responders. The solid horizontal line represents the median, the lower boundary represents the lower quartile, the upper border represents the upper quartile, and the whiskers represent outliers (t-test, **P < 0.01). (G) Box-and-whisker plot comparing the proportion of CD8+ T cells detected by in silico detection among healthy controls, IST responders, and non-responders. The solid horizontal line represents the median, the lower boundary represents the lower quartile, the upper border represents the upper quartile, and the whiskers represent outliers (t-test, *P < 0.05). BM = bone marrow; CLP = common lymphoid precursor; IST = immunosuppressive therapy; NK = natural killer cell; Treg = regulatory CD4+ T cell; UMAP = uniform manifold approximation and projection.
Depletion of MALAT1-upregulated Tregs in AA patientsNext, we investigated the characteristics of Tregs in AA patients and found that Tregs showed the pattern of depletion in AA patients (Figure 5A). The depletion was confirmed by flow cytometry, which measures the proportion of CD4+ Tregs from BM (Figure 5B,C). Importantly, we observed that 1 (C5) of the 2 Treg subclusters was characterized by significant depletion in AA patients compared with healthy controls (Figure 5D,E). To investigate the transcriptomic profile of the C5, we compared DEGs between the 2 Treg subclusters and found that MALAT1 was upregulated in C5 compared with the other Treg subcluster (C7) (Figure 5F; Suppl. Figure S7B). In addition, MALAT1high Tregs showed significant downregulation of FOS and JUNB, which are the genes inducing inflammatory responses in T cells (Figure 5F; Suppl. Figure S7B). Moreover, we verified the depletion of MALAT1high Tregs in AA patients using the independent scRNA-seq data of AA patients obtained from the Zhu et al.8 We identified MALAT1high Treg subcluster from Zhu et al8 based on the Pearson’s coefficient between the expression profiles of the T cells from Zhu et al8 and mean expression profiles of T cell subclusters from our data. The proportion of MALAT1high Treg subcluster from Zhu et al was similar to that of AA patients from our data, representing significant depletion compared with healthy controls from our data (Figure 5E). Additionally, we verified the depletion of MALAT1high Tregs by observing the proportion of MALAT1high Tregs from the integrated scRNA-seq data of healthy BM downloaded from the Human Cell Atlas37 and AA patients from our data after removing batch effect using Harmony.16 We reclustered T cells into 15 subclusters: 5 CD4+ T cell (C0, C1, C2, C5, C7), 2 Treg (C6, C8), 3 CD8+ T cell (C4, C10, C11), 2 NK cell (C3, C9, C12), and 2 common lymphoid precursor (CLP; C13, C14) subclusters (Figure 5G,H). The characteristic of each subcluster was identified based on the results of trajectory analysis (Suppl. Figure S8). We confirmed that the Tregs are significantly depleted in AA patients compared with Human Cell Atlas controls (Figure 5I). Especially, 1 (C8) of 2 Treg subclusters (C6, C8) was characterized with the significant depletion in AA patients and upregulation of MALAT1 compared with the other Treg subcluster (C8) (Figure 5J–F).
 Figure 5.:
Figure 5.: MALAT1 high Treg subcluster. (A) Box-and-whisker plot comparing the proportion of Tregs including C5 and C7 between healthy controls and AA patients. The solid horizontal line represents the median, the lower boundary represents the lower quartile, the upper border represents the upper quartile, and the whiskers represent outliers (t-test). (B) Representative images for CD4+ Tregs using flow cytometry. (C) Box-and-whisker plot comparing the percent of CD4+ Tregs among healthy controls, IST responders, and IST non-responders. The solid horizontal line represents the median, the lower boundary represents the lower quartile, the upper border represents the upper quartile, and the whiskers represent outliers (t-test, **P < 0.01 and *P < 0.05). (D) Box-and-whisker plot comparing the proportion of Treg subcluster C7 between healthy controls and AA patients. The solid horizontal line represents the median, the lower boundary represents the lower quartile, the upper border represents the upper quartile, and the whiskers represent outliers (t-test). (E) Box-and-whisker plot comparing the proportion of Treg subcluster C5 among healthy controls and AA patients from our data, and AA patients from Zhu et al. The solid horizontal line represents the median, the lower boundary represents the lower quartile, the upper border represents the upper quartile, and the whiskers represent outliers (t-test, **P < 0.01). (F) Volcano plot for DEGs between T cell subclusters 5 and 7. Red and blue dots denote upregulated and downregulated DEGs, respectively, with an adjusted P value <0.05 and a |log2(FC: fold change)| >0.25. (G) UMAP plot of T cells. (H) Heatmap of the expression of marker genes used to identify the cell type of each subcluster. Red and blue colors denote upregulated and downregulated genes, respectively. (I) UMAP plots displaying the scores associated with immune-suppressive potential of Tregs. (J) Relative expression profiles of MALAT1 among 10 T cell subclusters. (K) Box-and-whisker plot comparing the proportion of cluster 7 of clusters among HCA controls, healthy controls, and AA patients. The solid horizontal line represents the median, the lower boundary represents the lower quartile, the upper border represents the upper quartile, and the whiskers represent outliers (t-test, *P < 0.05). AA = aplastic anemia; BM = bone marrow; DEG = differentially expressed genes; HCA = Human Cell Atlas; IST = immunosuppressive therapy; UMAP = uniform manifold approximation and projection.
Depletion of activated memory B cells in IST non-respondersAfter reclustering B cells, 10 subclusters were identified: 1 immature B cell (C6), 4 mature B cells (C0, C1, C3, C4), 2 memory B cells (C2, C5), 1 transitional B cell (C9), and 2 plasma cells (C7, C8) (Figure 6A). The B cell subtypes were labeled according to the expression patterns of major B cell lineage markers (Figure 6B; Suppl. Table S9). The B cell subclusters were largely divided into 2 memory statuses based on the expression of CD27,38 while the expression patterns of IGHG1, IGHG4, and IGLL1 clearly distinguished plasma cells (C6, C9) from the other B cell subclusters (Figure 6A,B; Suppl. Figure S9). Interestingly, 1 memory B cell subcluster (C2) with relative upregulation of MALAT1, which was reported to be associated with viability of diffuse large B cell lymphoma (DLBCL) cell,39 and TXNIP, which is required for expansion of germinal center B cell,40 showed marginally significant depletion in IST non-responders (P = 0.058; Figure 6C–E).
 Figure 6.:
Figure 6.: B cell subclusters. (A) UMAP plot of B cells. (B) Heatmap of the expression of marker genes used to identify the cell type of each subcluster. Red and blue colors denote upregulated and downregulated genes, respectively. (C) Proportion of B cell subclusters in each sample. (D) Box-and-whisker plot comparing the proportion of B cell subcluster 0 among healthy controls, IST responders, and IST non-responders. The solid horizontal line represents the median, the lower boundary represents the lower quartile, the upper border represents the upper quartile, and the whiskers represent outliers (t-test, *P < 0.05). (E) Volcano plot for DEGs between B cell subclusters 2 and 5. Red and blue dots denote upregulated and downregulated DEGs, respectively, with an adjusted P value <0.05 and a |log2(FC: fold change)| >0.25. DEG = differentially expressed genes; IST = immunosuppressive therapy; UMAP = uniform manifold approximation and projection.
Depletion of CD1C+ DCs and M2 monocytic precursors, and enrichment of inflammatory M1 macrophages in AA patientsWhen reclustering myeloid cells, we identified 9 subclusters: 1 myeloid NK cells (C0), 1 myeloid NK precursors (C6),41,42 1 M1 macrophage (C7), 1 M1 monocyte (C8), 1 M2 monocyte (C4), 2 monocyte precursors (C1, C2), 1 CD1C+ DC (C3), and 1 CLEC9A+ DC (C5) (Figure 7A–C; Suppl. Table S10).43 Among the 9 subclusters, only 1 subcluster (C7) was marginally increased in AA patients (P = 0.055; Figure 7D,E; Suppl. Figure S10A). C7 was defined as inflammatory M1 macrophage based on the expression of FCGR1A, SOCS1, and complement components (C1QA, C1QB, and C1QC) (Figure 7B,F; Suppl. Figure S10B). In contrast, C3 and C4 were significantly depleted in AA patients and identified as CD1C+ DCs43 and M2 monocytic precursor, respectively, based on the expression profiles of CD1C and CLEC10A (Figure 7B,D–F; Suppl. Figure S10). To further explore the lineages of myeloid cells, we performed trajectory analysis and observed 2 different developmental branches of cellular differentiation (Figure 7C). One branch was from NK precursors (C6) to mature NK cells (C0), and the other was from monocyte precursors (C1, C2) to intermediate-stage M2 monocyte (C4), M1 monocyte (C8), and M1 macrophage (C7) (Figure 7C; Suppl. Figure S11). Late-stage M1 macrophage (C7) was enriched in AA patients, but late-stage CD1C+ DC (C3) and intermediate-stage M2 monocyte (C4) were depleted in AA patients (Figure 7E).
 Figure 7.:
Figure 7.: Myeloid cell subclusters. (A) UMAP plots of all myeloid cell subclusters (NK, DC; left panel) divided by healthy contr
留言 (0)