
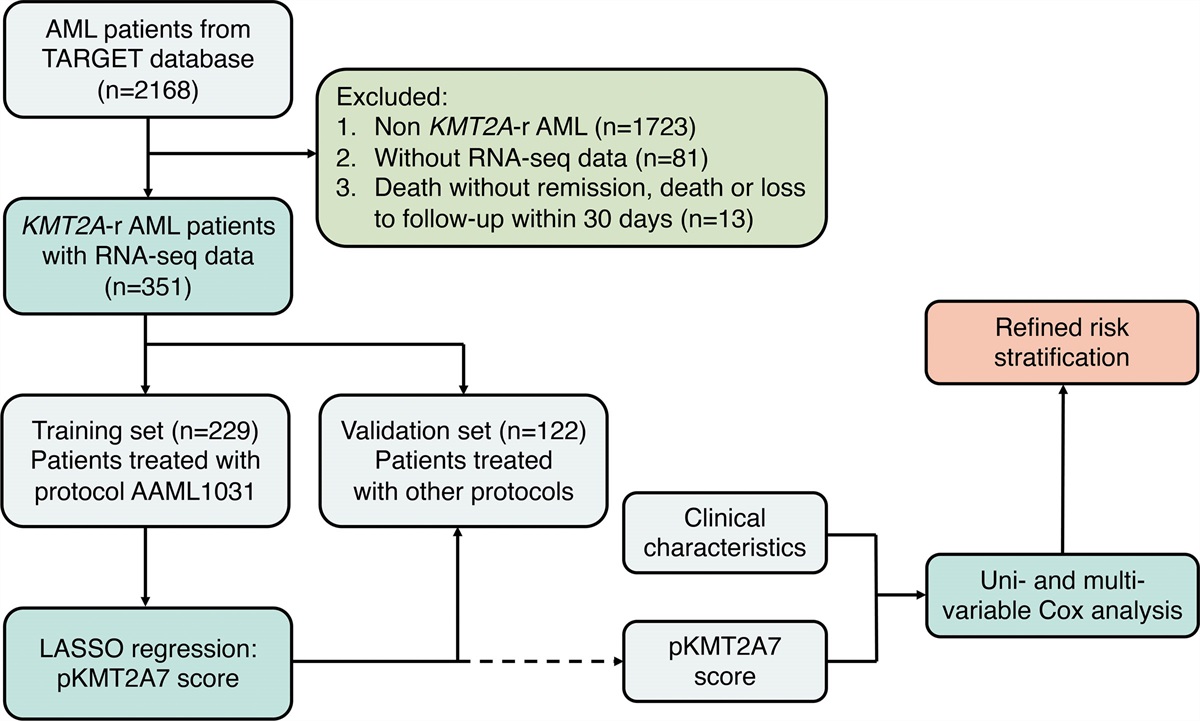
記住我



Inborn errors of metabolism (IEM) are a group of rare, heterogenous diseases caused mostly by monogenic mutations that result in disruption of metabolic pathways due to deficient enzymes, membrane transporters, or other functional regulators. The clinical features are shown by the accumulation of a toxic substrate, by deficiency of a product, or both.1 Most diseases are distinguished by multiorgan involvement, leading to progressive and life-threatening syndromes.2
Therapeutic approaches, enzyme replacement therapy (ERT), and allogeneic hematopoietic stem cell transplantation (HSCT)3 are directed toward the restoration of the equilibrium between substrate production and cleavage.4
ERT is now considered the standard-of-care for some IEM but, despite good results in slowing down disease progression, has many limitations: it is unable to reach the central nervous system (CNS) and the skeleton; it can induce the production of antibodies5; and it is expensive and not widely available. An alternative strategy to provide a long-lasting source of the missing protein is HSCT. The principle of HSCT for lysosomal storage disorders (LSD), a group of IEM, lies in cross-correction: engrafted donor-derived myeloid cells continually produce the missing enzyme, which is taken up by enzyme-deficient host cells.6 Moreover, the donor hematopoietic stem progenitor cells (HSPC) can migrate to the CNS and differentiate into resident macrophages (microglia) that secrete enzyme to the surrounding cells, improving neurocognitive outcomes.7 Currently, HSCT is the gold standard for mucopolysaccharidosis (MPS) I Hurler (MPSIH) patients younger than 2.5 years with mild neurological impairment8 and for X-linked adrenoleukodystrophy (X-ALD) patients with Loes score ≤9 and Neurologic Function Score ≤1.9 HSCT has been also tested in other IEM, such as metachromatic leukodystrophy (MLD) and Krabbe disease, but not all IEM seem to benefit from it possibly because normal enzyme expression obtained with healthy donor HSPC is not enough to achieve adequate systemic and local delivery of the enzyme, leaving the patients with a significant disease burden.10
In this perspective, HSPC-gene therapy (GT) represents a novel and promising alternative strategy11 based on the harvest and purification of autologous CD34+ HSPC transduced ex vivo with a viral vector to overexpress the therapeutic gene. After a chemotherapy-based conditioning regimen, the gene-modified cells are reinfused into the patient, where they can engraft and restore the missing protein production up to supraphysiologic levels (Figure 1).
 Figure 1.:
Figure 1.: HSPC-GT for IEM. Autologous HSPC are collected from the patient through bone marrow harvest or peripheral blood stem cell mobilization, and CD34+ cells are immunomagnetically selected and cultivated with the therapeutic vector. After the missing gene has been transferred to the HSPC, transduced cells are usually cryopreserved to wait for quality control release. Thereafter, the cells are thawed and reinfused into the patient after a chemotherapy-based preparative regimen. Once engrafted, the genetically modified HSPC will give rise to a corrected progeny, which will circulate and deliver the missing enzyme to different tissues and organs via the cross-correction mechanism. GT = gene therapy; HSPC = hematopoietic stem cells; IEM = inborn errors of metabolism.
To date, clinical trials have been conducted for the treatment of different IEM, including MLD, X-ALD, MPSIH, San Filippo syndrome (MPSIIIA), Gaucher and Fabry disease (Table 1), while 2 of these HSPC-GT have been made available on the European or US market.12,13
Table 1 - Ex Vivo Hematopoietic Stem Progenitor Cell-gene Therapy Clinical Trials in Inborn Errors of Metabolism Disease Study Name/ABCD1 = ATP-binding cassette, subfamily D, member 1; AGA = alpha-galactosidase A; ApoE2 = apolipoprotein E2; ARSA = arylsulfatase A; cDNA = complementary deoxyribonucleic acid; HSC = hematopoietic stem cells; IDS = iduronate-2-sulphatase; IDUA = alpha-L-iduronidase; LV = lentiviral vector; MLD = metachromatic leukodystrophy; MPS = mucopolysaccharidosis; MPSIH = mucopolysaccharidosis type I Hurler; PGK = phosphoglycerate kinase; SGSHX-ALD = X-linked adrenoleukodystrophy.
MLD is caused by the deficiency of the arylsulfatase A (ARSA) enzyme due to mutations in ARSA gene. This causes the accumulation of sulfatides in central and peripheral nervous system, leading to progressive demyelination and neurodegeneration. Three variants are identified based on the age of onset of first symptoms: late infantile (LI), juvenile (early [EJ] and late, before or after 7 years old, respectively), and adult.14 Especially for early-onset variants, ERT and HSCT have shown limited efficacy; therefore, an innovative approach became evident. Preclinical studies in MLD mice showed superiority of HSPC-GT over HSCT from wild-type donors, underlying the relevance of overexpressing the target gene to improve cross-correction and control neurological manifestations.15,16 Given these promising results, a phase I/II clinical trial based on HSPC transduced ex vivo with a lentiviral vector (LV) encoding ARSA in 9 LI and EJ patients at presymptomatic or very early-symptomatic stage was conducted17,18 achieving supranormal ARSA levels in circulating hematopoietic cells and normal enzyme in the cerebrospinal fluid (CSF), preserving cognitive function and severe motor impairment.15 Subsequently, a larger cohort of patients confirmed the same results in terms of safety and efficacy up to 7.5 years after HSPC-GT.18,19 In 2020, the European Medicine Agency granted marketing authorization for MLD HSPC-GT under the name of Libmeldy for LI presymptomatic and EJ pre- and early-symptomatic patients.12
X-ALD is a peroxisomal disease caused by a defect in the gene ABCD1 (ATP-binding cassette, subfamily D, member 1), which encodes a peroxisomal transporter (ALDP), resulting in abnormal breakdown of very long-chain fatty acids with consequent damage in adrenal and nervous system tissues.20 By adulthood, approximately half of the patients develops cerebral ALD (CALD), a severe inflammatory, demyelinating, and progressive condition leading to premature death.21 So far, HSCT has been the only effective therapy, when performed at an early-stage disease. The mechanisms responsible for its benefit is still unclear, as ALDP is not a soluble enzyme and in contrast with LSD, there is no cross-correction of neuronal cells. A possible mechanism could be held by donor-derived replacement of myeloid-derived cells in CNS. HSPC-GT was first tested in 2 boys with CALD and resulted in ABCD1 transgene expression in myeloid cells, metabolic correction, and clinical stabilization.22 Thereafter, 2 clinical trials (ALD-102 and ALD-104) using Lenti-D LV-modified HSPC saw equivalent results, where patients remained free of major functional disabilities and had attenuated progression of brain lesions.21 Following these results, HSPC-GT for CALD has been approved both in US23 and Europe with the aim to slow the progression of neurologic dysfunction in boys 4–17 years of age with early active CALD; however, it was withdrawn from the EU market for commercial reasons. While HSPC-GT and HSCT seem equally effective in preventing disease progression in patients with early-stage CALD and HSPC-GT is known to be associated with reduced transplant-related complications, 3 cases of myelodysplasia (MDS) have been recently reported after HSPC-GT.22,24
MPS are a group of LSD caused by deficiency in glycosaminoglycans (GAG) breakdown. The clinical manifestations include somatic and neurological symptoms, depending on the type of accumulated GAG.25 The severe form of MPSI (MPSIH), caused by the lack of alpha-L-iduronidase (IDUA) activity, is characterized by skeletal abnormalities, hepatosplenomegaly, hearing and visual loss, cardiac and pulmonary problems, and progressive cognitive impairment. With ERT affected individuals die in the first decade of life.8 Another form of MPS is MPS IIIA, caused by deficiency of N-sulfoglucosamine sulfohydrolase (SGSH) with accumulation of heparan sulfate. Children with severe MPSIIIA present progressive neurodegeneration with premature death.26 While HSCT represents standard-of-care for MPSIH and has significantly improved survival, residual disease burden persists in the long-term especially at the level of the CNS and skeleton. Preclinical studies in mouse models of both MPSIH and MPSIIIA treated with LV-based HSPC-GT have shown superior efficacy over HSCT.27,28 Interim results of the first-in-human phase I/II clinical trial of HSPC-GT with IDUA-LV showed supraphysiologic blood IDUA activity with early normalization or near-normalization of GAG excretion in 8 patients. IDUA activity became detectable in CSF after treatment and was associated with local clearance of GAG. In parallel, patients experienced stable cognitive and motor performances, improved joint stiffness, and normal growth.29 Concerning MPSIIIA, a phase I/II clinical trial of HSPC-GT using LV-SGSH has enrolled 5 patients with promising preliminary results in term of safety and efficacy, supraphysiologic blood SGSH activity, and GAG depletion.30 For MPSII,31 a HSPC-GT trial was recently opened at Manchester University for patients between 3 and 12 months of age who will be treated with HSPC transduced ex vivo with a LV encoding human iduronate-2-sulphatase (IDS) gene with an ApoE2 tag to improve neurological correction,32 increasing further the range of action of HSPC-GT in MPS.
HSPC-GT has also been investigated for the treatment of Fabry disease, a X-linked condition caused by alpha-galactosidase A (Gal A) deficiency with consequent glycosphingolipids accumulation and neurological symptoms.33 Two clinical trials of HSPC-GT have been conducted with preliminary results showing efficient LV-mediated gene transfer, increased intracellular and circulating Gal A activity. Nevertheless, enzyme levels declined over time, underlying poor engraftment34 and the company developing the product announced the deprioritization of Fabry program removing it from its pipeline.
Another sphingolipidoses, Gaucher type 1 disease (GD1), is caused by mutation in glucosidase beta acid 1 (GBA1) gene, resulting in insufficient activity of the enzyme glucocerebrosidase, with progressive accumulation of glucocerebroside and several clinical manifestations, including cytopenia, hepatosplenomegaly, and skeletal alterations. After demonstration of HSPC-GT efficacy in preventing disease progression and in the long-term improving hepatosplenomegaly and blood counts in GD1 mouse model,35,36 recently a clinical trial based on autologous HSPC genetically modified with a LV encoding for human glucocerebrosidase has been initiated. A Gaucher type 3 disease (GD3) patient has been recently treated on a named patient basis with HSPC-GT at Manchester University with initial biomarker and clinical response (R. Wynn, personal communication). A clinical trial of HSPC-GT for GD3 is expected to open soon.
STRENGTHS AND SHORTCOMINGS IN HSPC-GT FOR IEM Strengths Reduced transplant-related complications and mortalityPotential complications of HSCT are represented by the rejection of donor graft and graft versus host disease (GvHD), caused by the immunological differences between patient and donor.37 HSPC-GT is based on the use of autologous cells, which minimizes the risk of rejection and GvHD, not requiring immunosuppression after transplantation.38 Other risks associated with HSCT include conditioning-related toxicity and infections. While the intensity of chemotherapy in IEM may not be different from that of HSCT, the use of autologous cells in HSPC-GT allows early reconstitution of innate and adaptive immunity, leading to a low incidence of posttransplantation infectious complications, despite lympho- and myeloablation.19,29
LSD patients may develop autoantibodies against the enzyme/transgene after HSPC-GT, which may potentially hamper its efficacy. However, the transient development of anti-ARSA antibodies in 14% of the early-onset MLD patients after HSPC-GT19 resolved spontaneously or after B-cell depleting therapy and did not impact on clinical and safety outcomes. ERT-induced anti-IDUA IgG antibodies, present in 5 of the 7 MPSIH patients before HSPC-GT, disappeared in the first 3 months after treatment29 likely due to the lymphodepleting conditioning, which may have helped inducing tolerance.39
Overall, HSPC-GT holds a favorable safety profile requiring short hospitalization due to prompt engraftment, with very limited transplant-related complications.24 This, in turn, could positively impact on neurological and skeletal functions of patients affected by IEM.40
Donor availability and timely transplantationAnother significant advantage of using autologous cells is that there is no need of searching for a suitable donor, while HSCT frequently depends on the availability of unrelated compatible donors, as potentially HLA-matched siblings or family members are often disease carriers, not candidates as donors. Thus, employing autologous HSPC allows timely transplantation, which is crucial for long-term clinical outcome in IEM.24 Precocious recognition of these disorders by newborn screening (NBS) could enable early diagnosis41 and treatment at presymptomatic, currently possible only for patients with a positive family history, further optimizing clinical results with HSPC-GT strategies. In addition, the development of low toxicity conditioning regimens based on monoclonal antibodies selectively depleting blood cells in the bone marrow might encourage the application of HSPC-GT in the neonatal age.42,43
Overexpression of the therapeutic enzymeIn HPSC-GT strategies for LSD, autologous cells are genetically modified to constitutively express supranormal levels of a therapeutic enzyme and become a quantitatively more effective source of functional enzyme if compared with HSCT.44,45 The supraphysiological enzyme levels produced by the gene-corrected HSPC progeny, circulating or resident in a specific tissue, allows cross-correction of nonhematopoietic cells, including neurons and skeletal cells (Figure 1). This has been postulated in MLD where microglia replacement by the gene-modified myeloid cells provides an effective local source of bioavailable enzyme capable to clear the accumulated sulfatides.17,18 In support of skeletal correction, in vitro data show that gene-modified CD34+-derived osteoclasts from MPSIH patients after HSPC-GT secrete supraphysiologic levels of functional IDUA enzyme, compared with healthy donor cells, which could represent a source of enzyme for resident skeletal cells involved in altered bone remodeling.46–48 Other target organs, such as the eye and the heart, require overexpression of the transgene for optimal clinical outcome. See also Table 2.
Table 2 - Strengths and Shortcomings of Hematopoietic Stem Progenitor Cell-gene Therapy for Inborn Errors of Metabolism Strengths Absent risk for graft rejection and GvHDGvHD = graft versus host disease; HSPC = hematopoietic stem cells; MLD = metachromatic leukodystrophy; MPS IH = mucopolysaccharidosis type I Hurler; NBS = newborn screening.
A potential risk of HSPC-GT is related to vector-mediated insertional mutagenesis. This is due to the possibility that the insertion site of viral genome would lie close to an oncogene, altering the expression patterns and leading to the development of MDS or leukemia. Insertional mutagenesis has been reported with gamma retroviral vectors (RVs) inducing leukaemia/lymphomas.49 Overall, factors that may aggravate the genotoxic risk of vector integration are the vector design, the promoter choice, and the quality of manufactured cell product.50 LV can reduce the genotoxicity compared with RV by using a vector internal promoter to drive transcription of the therapeutic transgene and the self-inactivating design.51–53 Overall, the safety and efficacy data generated in LV-based clinical trials19,29,54–58 support its use to treat patients affected by IEM.
Nevertheless, MDS was diagnosed in 3 patients treated with LV-based HSPC-GT for CALD within the ALD-102 and ALD-104 studies, 14 months, 25 months, and 7.5 years after treatment, respectively. According to the current evidence, the development of MDS was mediated by Lenti-D LV insertion. Specific design features of this vector, such as the presence of the MNDU3 promoter enabling ubiquitous expression of the ABCD1 transgene in blood lineage, likely contributed to the development of MDS.24 However, after having placed a clinical hold on this product, on September 2022, FDA approved it with a boxed warning for hematologic malignancy, including life-threatening cases of MDS, based on the cost-benefit analysis.23
Limited follow-upSo far, the clinical trials developed for IEM have not reached a follow-up long enough to draw definitive conclusions on safety and efficacy. A longer observation is required to monitor for potential side effects of HSPC-GT, such as insertional mutagenesis abovementioned,59 and verify long-term clinical efficacy and clinical relevance of previous damage accumulated by storage substrates. While evidence of clinical efficacy of HSPC-GT in MLD is available up to 7.5 years after treatment,19 whether delivering supranormal quantities of a missing protein in the target tissues represents a superior therapeutic strategy over standard of care needs to be confirmed in the other LSD.
High production costsRecent news of disinvestment in HSPC-GT for rare genetic defects are partially explained by high production costs and difficulties in achieving market access and requested reimbursement.60 HSPC-GT requires high investment due to elevated costs of the starting materials, including viral vectors, the manufacturing in dedicated Good Manifacturing Practice facilities, and together with the complex monitoring of patients for long term after the treatment. These expenditures cannot be easily compensated in the setting of rare diseases or when other treatment options are already available. Moreover, the long and cumbersome pathway to reach market access in terms of regulatory requirement poses further challenges to the development of these products.60 See also Table 2.
CONCLUSIONS AND FUTURE PERSPECTIVESo far, encouraging results have been achieved by completed and ongoing clinical trials of HSPC-GT in IEM, highlighting HSPC-GT ability to achieve metabolic correction and clinical efficacy in MLD, X-ALD, and preliminarly in MPSIH and MPSIII.9,19,29,30 This has led to the marketing authorization of 2 of these products.12,13
Nevertheless, the recent MDS cases in CALD patients after HSPC-GT enhance the need of further evaluating long-term safety. In addition, the limited follow-up in some diseases hampers a proper comparison between HSPC-GT and standard of care, that is, ERT and HSCT.
The potential efficacy of HSPC-GT in correcting LSD-related neurological and skeletal manifestations opens the door for the development of similar strategies in other rare IEM with an unmet medical need. Ideal candidates could be LSD characterized by neurological impairment and/or skeletal dysplasia for whom efficacious therapeutic options are currently not available, such as other MPS (ie, MPSIV and MPSVI), alpha mannosidosis, sialidosis, mucolipidosis type III, gangliosidosis GM1, pycnodisostosis, and Niemann-Pick disease. Leveraging on the biochemical and clinical results obtained so far in MLD, MPSIH, and MPSIIIA, it is meaningful to invest on innovative platform approaches to move from a single disease to a simultaneous multidisease model. Given the high costs for the development of such therapies together with the long regulatory procedures, moving to a platform approach could help reducing time and costs making them more sustainable and accessible to patients.60,61
Notably, the implementation of NBS programs will enable early diagnosis, identification of ideal candidate patients for HSPC-GT, and timely treatment.
Modification of defective genes by site-specific in vivo genome editing is being explored in preclinical studies and clinical trials with promising results.62,63 It is likely that in the future, this innovative technology will contribute to further implement the therapeutic options for IEM.
AUTHOR CONTRIBUTIONSFT, GC, and MC wrote the article. MEB revised it and approved its final version. All authors edited and approved the article for submission and publication.
DISCLOSURESThe authors have no conflicts of interest to disclose.
SOURCES OF FUNDINGThe authors declare no sources of funding.
REFERENCES 1. Ezgu F. Inborn errors of metabolism. Adv Clin Chem. 2016;73:195–250. 2. Boustany RM. Lysosomal storage diseases--the horizon expands. Nat Rev Neurol. 2013;9:583–598. 3. Platt FM, Jeyakumar M. Substrate reduction therapy. Acta Paediatr. 2008;97:88–93. 4. Fernandez-Pereira C, San Millán-Tejado B, Gallardo-Gómez M, et al. Therapeutic approaches in lysosomal storage diseases. Biomolecules. 2021;11:1775. 5. Broomfield A, Jones SA, Hughes SM, et al. The impact of the immune system on the safety and efficiency of enzyme replacement therapy in lysosomal storage disorders. J Inherit Metab Dis. 2016;39:499–512. 6. Neufeld EF, Muenzer J, Scriver C, Beaudet A, Sly W, Valle D. The Metabolic and Molecular Basis of Inherited Disease (ed NY McGraw-Hill). 2001;3421–3452. 7. Tan EY, Boelens JJ, Jones SA, et al. Hematopoietic stem cell transplantation in inborn errors of metabolism. Front Pediatr. 2019;7:433. 8. Muenzer J, Wraith JE, Clarke LA, International Consensus Panel on Management and Treatment of Mucopolysaccharidosis I. Mucopolysaccharidosis I: management and treatment guidelines. Pediatrics. 2009;123:19–29. 9. Eichler F, Duncan C, Musolino PL, et al. Hematopoietic stem-cell gene therapy for cerebral adrenoleukodystrophy. N Engl J Med. 2017;377:1630–1638. 10. Chiesa R, Wynn RF, Veys P. Haematopoietic stem cell transplantation in inborn errors of metabolism. Curr Opin Hematol. 2016;23:530–535. 11. Nagree MS, Scalia S, McKillop WM, et al. An update on gene therapy for lysosomal storage disorders. Expert Opin Biol Ther. 2019;19:655–670. 14. van Rappard DF, Boelens JJ, Wolf NI. Metachromatic leukodystrophy: disease spectrum and approaches for treatment. Best Pract Res Clin Endocrinol Metab. 2015;29:261–273. 15. Biffi A, Capotondo A, Fasano S, et al. Gene therapy of metachromatic leukodystrophy reverses neurological damage and deficits in mice. J Clin Invest. 2006;116:3070–3082. 16. Biffi A, De Palma M, Quattrini A, et al. Correction of metachromatic leukodystrophy in the mouse model by transplantation of genetically modified hematopoietic stem cells. J Clin Invest. 2004;113:1118–1129. 17. Biffi A, Montini E, Lorioli L, et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science. 2013;341:1233158. 18. Sessa M, Lorioli L, Fumagalli F, et al. Lentiviral haemopoietic stem-cell gene therapy in early-onset metachromatic leukodystrophy: an ad-hoc analysis of a non-randomised, open-label, phase 1/2 trial. Lancet. 2016;388:476–487. 19. Fumagalli F, Calbi V, Natali Sora MG, et al. Lentiviral haematopoietic stem-cell gene therapy for early-onset metachromatic leukodystrophy: long-term results from a non-randomised, open-label, phase 1/2 trial and expanded access. Lancet. 2022;399:372–383. 20. Moser HW. Adrenoleukodystrophy: phenotype, genetics, pathogenesis and therapy. Brain. 1997;120(Pt 8):1485–1508. 21. Gupta AO, Raymond G, Pierpont EI, et al. Treatment of cerebral adrenoleukodystrophy: allogeneic transplantation and lentiviral gene therapy. Expert Opin Biol Ther. 2022;22:1151–1162. 22. Cartier N, Hacein-Bey-Abina S, Bartholomae CC, et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science. 2009;326:818–823. 24. Chiesa R, Bernardo ME. Haematopoietic stem cell gene therapy in inborn errors of metabolism. Br J Haematol. 2022;198:227–243. 25. Platt FM, d’Azzo A, Davidson BL, et al. Lysosomal storage diseases. Nat Rev Dis Primers. 2018;4:27. 26. Seker Yilmaz B, Davison J, Jones, Baruteau SA. Novel therapies for mucopolysaccharidosis type III. J Inherit Metab Dis. 2021;44:129–147. 27. Visigalli I, Delai S, Politi LS, et al. Gene therapy augments the efficacy of hematopoietic cell transplantation and fully corrects mucopolysaccharidosis type I phenotype in the mouse model. Blood. 2010;116:5130–5139. 28. Sergijenko A, Langford-Smith A, Liao AY, et al. Myeloid/microglial driven autologous hematopoietic stem cell gene therapy corrects a neuronopathic lysosomal disease. Mol Ther. 2013;21:1938–1949. 29. Gentner B, Tucci F, Galimberti S, et al. Hematopoietic stem- and progenitor-cell gene therapy for hurler syndrome. N Engl J Med. 2021;385:1929–1940. 30. Wynn RF, Kinsella JL, Holley RJ, et al. Biochemical engraftment and clinical outcomes following ex-vivo autologous stem cell gene therapy for mucopolysaccharidosis type IIIA. Blood. 2022;140:1897–1898. 31. Sestito S, Falvo F, Scozzafava C, et al. Genetics and gene therapy in hunter disease. Curr Gene Ther. 2018;18:90–95. 32. Gleitz HF, Liao AY, Cook JR, et al. Brain-targeted stem cell gene therapy corrects mucopolysaccharidosis type II via multiple mechanisms. EMBO Mol Med. 2018;10:e8730. 33. Miller JJ, Kanack AJ, Dahms NM. Progress in the understanding and treatment of Fabry disease. Biochim Biophys Acta Gen Subj. 2020;1864:129437. 34. Khan A, Barber DL, Huang J, et al. Lentivirus-mediated gene therapy for Fabry disease. Nat Commun. 2021;12:1178. 35. Dahl M, Smith EMK, Warsi S, et al. Correction of pathology in mice displaying Gaucher disease type 1 by a clinically-applicable lentiviral vector. Mol Ther Methods Clin Dev. 2021;20:312–323. 36. Enquist IB, Nilsson E, Månsson J-E, et al. Successful low-risk hematopoietic cell therapy in a mouse model of type 1 gaucher disease. Stem Cells. 2009;27:744–752. 37. DeFilipp Z, Hefazi M, Chen YB, et al. Emerging approaches to improve allogeneic hematopoietic cell transplantation outcomes for nonmalignant diseases. Blood. 2022;139:3583–3593. 38. Morgan RA, Gray D, Lomova A, et al. Hematopoietic stem cell gene therapy: progress and lessons learned. Cell Stem Cell. 2017;21:574–590. 39. Squeri G, Passerini L, Ferro F, et al. Targeting a pre-existing anti-transgene T cell response for effective gene therapy of MPS-I in the mouse model of the disease. Mol Ther. 2019;27:1215–1227. 40. Parris KR, Russell KM, Triplett BM, et al. Neurocognitive functioning in long-term survivors of pediatric hematopoietic cell transplantation. Bone Marrow Transplant. 2021;56:873–882. 41. Matern D, Gavrilov D, Oglesbee D, et al. Newborn screening for lysosomal storage disorders. Semin Perinatol. 2015;39:206–216. 42. Czechowicz A, Palchaudhuri R, Scheck A, et al. Selective hematopoietic stem cell ablation using CD117-antibody-drug-conjugates enables safe and effective transplantation with immunity preservation. Nat Commun. 2019;10:617. 43. Palchaudhuri R, Saez B, Hoggatt J, et al. Non-genotoxic conditioning for hematopoietic stem cell transplantation using a hematopoietic-cell-specific internalizing immunotoxin. Nat Biotechnol. 2016;34:738–745. 44. Aldenhoven M, Jones SA, Bonney D, et al. Hematopoietic cell transplantation for mucopolysaccharidosis patients is safe and effective: results after implementation of international guidelines. Biol Blood Marrow Transplant. 2015;21:1106–1109. 45. Aldenhoven M, Wynn RF, Orchard PJ, et al. Long-term outcome of Hurler syndrome patients after hematopoietic cell transplantation: an international multicenter study. Blood. 2015;125:2164–2172. 46. Gatto F, Redaelli D, Salvadè A, et al. Hurler disease bone marrow stromal cells exhibit altered ability to support osteoclast formation. Stem Cells Dev. 2012;21:1466–1477. 47. Tomatsu S, Alméciga-Díaz CJ, Montaño AM, et al. Therapies for the bone in mucopolysaccharidoses. Mol Genet Metab. 2015;114:94–109. 48. Santi L, Crippa S, Capo V, et al. Skeletal damage and cross-correction in MPSIH HSPC-gene therapy. Hum Gene Ther. 2022;33:A4–A4. 49. Bushman FD. Retroviral insertional mutagenesis in humans: evidence for four genetic mechanisms promoting expansion of cell clones. Mol Ther. 2020;28:352–356. 50. Tucci F, Galimberti S, Naldini L, et al. A systematic review and meta-analysis of gene therapy with hematopoietic stem and progenitor cells for monogenic disorders. Nat Commun. 2022;13:1315. 51. Braun CJ, Boztug K, Paruzynski A, et al. Gene therapy for Wiskott-Aldrich syndrome--long-term efficacy and genotoxicity. Sci Transl Med. 2014;6:227ra233. 52. Siler U, Paruzynski A, Holtgreve-Grez H, et al. Successful Combination of Sequential Gene Therapy and Rescue Allo-HSCT in Two Children with X-CGD - Importance of Timing. Curr Gene Ther. 2015;15:416–427. 53. Ott MG, Schmidt M, Schwarzwaelder K, et al. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat Med. 2006;12:401–409. 54. Aiuti A, Biasco L, Scaramuzza S, et al. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science. 2013;341:1233151. 55. Ferrua F, Cicalese MP, Galimberti S, et al. Lentiviral haemopoietic stem/progenitor cell gene therapy for treatment of Wiskott-Aldrich syndrome: interim results of a non-randomised, open-label, phase 1/2 clinical study. Lancet Haematol. 2019;6:e239–e253. 56. Marktel S, Scaramuzza S, Cicalese MP, et al. Intrabone hematopoietic stem cell gene therapy for adult and pediatric patients affected by transfusion-dependent ss-thalassemia. Nat Med. 2019;25:234–241. 57. Ribeil JA, Hacein-Bey-Abina S, Payen E, et al. Gene therapy in a patient with sickle cell disease. N Engl J Med. 2017;376:848–855. 58. Kohn DB, Booth C, Shaw KL, et al. Autologous ex vivo lentiviral gene therapy for adenosine deaminase deficiency. N Engl J Med. 2021;384:2002–2013. 59. Howe SJ, Mansour MR, Schwarzwaelder K, et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J Clin Invest. 2008;118:3143–3150. 60. Aiuti A, Pasinelli F, Naldini L. Ensuring a future for gene therapy for rare diseases. Nat Med. 2022;28:1985–1988. 61. Fox T, Bueren J, Candotti F, et al. Access to gene therapy for rare diseases when commercialization is not fit for purpose. Nat Med. 2023;29:518–519. 62. Gomez-Ospina N, Scharenberg SG, Mostrel N, et al. Human genome-edited hematopoietic stem cells phenotypically correct mucopolysaccharidosis type I. Nat Commun. 2019;10:4045. 63. Frangoul H, Altshuler D, Cappellini MD, et al. CRISPR-Cas9 gene editing for sickle cell disease and beta-thalassemia. N Engl J Med. 2021;384:252–260.
留言 (0)