

記住我
Amyotrophic lateral sclerosis (ALS) is a rapidly progressive neurodegenerative disorder of the motor neurons. The site of onset is variable in ALS based on the phenotype, although a spinal/limb onset (58–82%) followed by bulbar onset (28%) is the classical phenotype (1). The spinal onset phenotype is further classified into typical spinal onset in which lower motor neuron (LMN) and upper motor neuron (UMN) symptoms start in one limb and rapidly spread to all other limbs, bulbar, and thoracic regions; flail arm and leg phenotypes in which predominantly LMN symptoms remain restricted to the upper or lower limbs, respectively, for at least 12 months; hemiplegic in which predominantly UMN symptoms remain restricted to ipsilateral upper and lower limbs; and psudopolyneuritic phenotype presenting with distal-predominant LMN findings. Other rare phenotypes include progressive muscular atrophy (PMA), primary lateral sclerosis (PLS), mixed, and thoracic/respiratory onset (2).
The short life of ALS patients is burdened by progressive loss of motor and sometimes cognitive abilities. Therefore, an enormous amount of healthcare services and resources are vital throughout this journey to support patients and their caregivers. These include but are not limited to regular neurologic evaluations, speech therapy, physical and occupational therapy, pulmonary function evaluation and interventions, nutritional interventions and enteral feeding, psychiatric evaluations, and palliative care. The worldwide solution that allows integrated care for these patients while also preventing escalated healthcare costs is multidisciplinary ALS clinics which are known as the gold standard of ALS care. It is a quality milestone for the Iranian healthcare system to introduce this standard to the care of Iranian ALS patients. To this end, a national ALS clinical practice guideline is the necessary first step.
2. MethodsA team of national neurology, neuromuscular, rehabilitation, pulmonology, speech-language pathology, psychiatry, legal and medical ethics, and nutrition experts participated to develop the Iranian ALS clinical practice guideline. Clinical questions were formulated in the Patient, Intervention, Comparison, and Outcome (PICO) format to guide the literature search. PICO questions were finalized in a panel discussion. Search strategies were developed based on the PICO questions, and relevant literature was retrieved from Medline, Embase, Cochrane Database of Systematic Reviews (CDSR), and Cochrane Central Register of Controlled Trials (CENTRAL) filtering for English and Persian language. Considering the current lack of adequate national/local studies, a consensus-based approach was taken to evaluate the quality of the retrieved evidence and formulate recommendations. Panel discussions were held to present the findings of the literature search and draft recommendations. The recommendations were finalized in separate panel discussions. We graded the strength of the recommendations based on experts' consensus on scientific rigor and quality of evidence, as well as feasibility measures including availability, cultural acceptance, specific legal aspects, and relevant costs. Grade A was applied when a highly feasible recommendation was supported by high-quality evidence, and grade B was applied when feasibility was lower or lower-quality evidence was available for a highly feasible recommendation. Grade C was applied when both evidence quality and feasibility were low, and grade E was applied to good clinical practice points solely based on expert opinion with any feasibility rating. The final guideline was reviewed and approved by all experts.
3. Results 3.1. EpidemiologyThe overall crude worldwide prevalence of ALS is approximately 4.42 per 100,000 (3), and it is estimated that by 2040 the Iranian ALS population will roughly reach 3,000 (4). The first epidemiological study of the Iranian ALS population was published in 2010 reporting on 98 ALS patients evaluated during a 4-year period in outpatient or inpatient departments of Isfahan Medical University located in central Iran (5). Comparing the findings of this study with the worldwide data in a meta-analysis (6) showed that the mean age of onset is lower among Iranian patients (<55 years) compared to Europe and New Zealand (63–65 years) as well as America and East Asia (59 years). In addition, the frequency of bulbar-onset phenotype in the Iranian study (27%) was lower compared to Europe (45% in the Northern areas to 34% in the Western and Southern areas) but closer to the East Asia (28%), Israel (22%), and America (28%) estimates. This finding, along with younger age of onset, might explain the higher survival reported in the Iranian study (48 months) compared to the other areas (25–30 months in Europe and 35 months in North America). Another difference was in the sex ratio (M/F) which was higher in the Iranian study (approximately 2) compared to Europe, North America, and New Zealand (1.22–1.33) but closer to Asia (1.55 in East and 1.72 in West Asia), Uruguay, Libya, and Hawaii (>2). In another study from Mashhad in Northeast Iran, 59 consecutive ALS patients were evaluated (7). A similar age of onset (48 years), sex ratio (1.8), and survival (53 months) were reported, while the bulbar-onset phenotype was even less frequent (15%). The rate of familial ALS was 3.8% in this study which is close to the worldwide estimate (4.7%) (6). Tracheostomy with mechanical ventilation was reported in 20% of patients in this sample. The largest natural history study of Iranian ALS patients, published in 2015, recruited 358 patients from 10 centers in the country (8). Findings were generally in agreement with the previous two reports with the mean age of onset being 52 years and the rate of bulbar-onset and familial ALS at 23% and 3.4%, respectively. The sex ratio was 1.6 which is closer to most other regions. Approximately 23% of patients died during a 12-month follow-up.
3.2. Clinical/electrodiagnostic criteriaDiagnosis of ALS is based on clinical judgment supported by the findings from a standard and comprehensive electrodiagnostic study. Such a study should include an evaluation of muscles with different nerve and root innervation in the proximal and distal of at least three limbs, three segments of thoracic paraspinal muscles, and at least one bulbar muscle. El Escorial World Federation of Neurology criteria for the diagnosis of ALS were published in 1994 (9) and revised later in 2000 to improve its sensitivity (10). Awaji criteria (AC) were subsequently proposed in 2008 to improve the integration of electrodiagnostic studies into the existing ALS diagnostic criteria aiming to further increase sensitivity (11). In fact, current evidence indicates that the Awaji criteria are significantly more sensitive than the revised El Escorial criteria (73 vs. 58%, respectively) (12–15).
Despite improved sensitivity, complexity and low inter-rater reliability were still major barriers to the use of the Awaji criteria in a multicenter study (16). To address these limitations, a consensus meeting led by the World Federation of Neurology was held in 2019 to develop a new set of criteria (17). The resulting Gold Coast criteria (Table 1) are believed to be simpler, more sensitive, and more useful in clinical settings than the previous criteria (18–20). Current improvements in the care of ALS patients, especially in the setting of multidisciplinary clinics, necessitate highly sensitive diagnostic criteria that also allow earlier diagnosis. However, it is important to note that, as part of the clinical criteria, ALS mimics and differential diagnoses should be excluded using appropriate evaluations before a diagnosis could be made.
Table 1. Gold coast ALS criteria.
3.2.1. Recommendations• The application of the Gold Coast criteria results in an early detection of ALS patients and is recommended for the diagnosis of ALS in clinical practice (grade A).
• The exclusion of ALS mimics is mandatory for the diagnosis of ALS in clinical settings (grade A).
3.3. Monitoring of disease progression 3.3.1. Clinical motor scaleThe ALS functional rating scale (ALSFRS) and its revised version, ALSFRS-R, are widely used survival predictors and outcome measures in ALS patients (21, 22). ALSFRS-R consists of 12 components on a 0 to 4 scale and has good inter-rater and intrarater reliability (23). Multiple studies have shown that ALSFRS-R can be used as a marker of disease progression with higher progression rates being indicative of lower survival (24–26). However, clinicians should be aware of the limitations of this scale which include relative insensitivity to change in short periods of time (especially under 6 months), the subjective nature of the scale, insensitivity to small changes especially in patients with less severe disease, and lack of unidimensionality which means that the scale should be considered as three separate subscales (bulbar, fine and gross motor, and respiratory) rather than a single scale (27–30).
3.3.2. Motor staging systemsAlthough multiple staging systems have been developed for the evaluation of various aspects of ALS, the most widely used systems are King's clinical staging and Milano-Torino (MiToS) functional staging systems (31, 32). Both are simple systems that can easily be applied in busy clinical settings. The MiToS system basically divides ALSFRS-R into four domains and counts the number of domains in which the patient has lost his/her independence. These domains include walking and self-care, swallowing, communicating, and breathing. A stage 0 will be applied when functional impairment has not caused loss of independence in any domain. In King's staging, stages 1–3 represent the number of body regions involved, including bulbar, upper limbs, and lower limbs. Stage 4 is when there is nutritional or respiratory failure necessitating gastrostomy or non-invasive ventilation (NIV). Stage 5 in both systems represents death. King's staging can also be derived from ALSFRS-R (33).
3.3.3. Measures of quality of lifeThe ALS Assessment Questionnaire (ALS-AQ40) is a health-related quality-of-life measure for ALS with high internal reliability and validity (34, 35). A Persian version is also available (36).
3.3.4. Cognitive/behavioral screensAs discussed in the cognitive section below, cognitive/behavioral dysfunction affects more than half of the patients with ALS, leading to frank dementia in approximately 15–20% (37). Cognitive dysfunction in ALS is associated with poor outcomes (38). Several cognitive/behavioral screens are available; however, no single tool has consistently been used in related studies (39–41). The Edinburgh Cognitive and Behavioral ALS Screen (ECAS) is a more comprehensive tool that evaluates a wide range of cognitive functions, including a detailed evaluation of language and social cognition domains. The ALS Cognitive Behavioral Screen (ALS-CBS) is a simpler tool that more specifically measures executive dysfunction (42). Both scales also evaluate behavioral changes based on a caregiver-administered questionnaire. A validated Persian version of the ECAS is currently available (43).
3.3.5. Objective measurement of motor functionThe Motor Unit Number Estimation (MUNE) and the Motor Unit Number Index (MUNIX) are quantitative neurophysiological measures that estimate the number of remaining motor units in a muscle. These methods provide a more sensitive method to quantitatively measure ALS progression compared to clinical methods even in pre-symptomatic muscles (44–48). These features make MUNE and MUNIX good biomarkers of disease progression in clinical research; however, clinical scales are more widely available and easy to use in clinical settings.
3.3.6. Recommendations• Use of ALSFRS-R as a measure of disability is recommended for all ALS patients at baseline and follow-up visits (grade A).
• ALS-CBS or ECAS is recommended for cognitive/behavioral screening of ALS patients at baseline and follow-up visits (grade A).
• Use of disease-specific quality-of-life assessment for ALS such as ALSAQ-40 is recommended in baseline and follow-up visits of all ALS patients in tertiary ALS clinics (grade B).
3.4. Differential diagnostic workupAdditional workups in ALS mostly focus on excluding differential diagnoses and constitute an important step in the diagnosis of ALS. Depending on the clinical presentations, various combinations of diagnostic tests might be considered in the evaluation of an individual patient.
3.4.1. Neuraxis neuroimagingSpinal cord magnetic resonance imaging (MRI) is essential to exclude the most common and important ALS mimics, spinal spondylosis, or other space-occupying lesions. Cervical spondylotic amyotrophy is a relatively rare form of myelopathy that can involve more proximal (C5, C6) or distal (C7, C8, and T1) cervical spinal segments (49). Cervical MRI may reveal T2 hyperintensity in addition to central and foramina canal stenosis in both types (50). On the other hand, comorbid cervical spondylosis and cervical cord compression are more common in ALS patients compared to other neurodegenerative and neuromuscular disorders (51). Spinal MRI also provides diagnostic clues in other ALS mimics, such as radiation myelopathy and Hirayama disease. In multifocal motor neuropathy, another treatable mimic of motor neuron disease (MND), contrast-enhanced MRI of the root and the brachial/lumbosacral plexus increases diagnostic certainty (52).
3.4.2. Serum vitamin B12A large retrospective chart review study showed that laboratory workup resulted in a change in management in 6% of patients but did not change the diagnosis of ALS in any patient. Assays with higher rates of abnormal findings included complete blood count, vitamin B12, serum creatine kinase, and parathyroid hormone (53).
A few case reports of patients with an ALS-like presentation but with a final diagnosis of B12 deficiency have been reported in the literature (54, 55). However, no cases of vitamin B12 deficiency were reported among ALS mimickers from the Irish and Scottish ALS registries (56, 57). Despite the rarity as an ALS mimicker, considering the treatable nature of the disease and the low cost of screening, it is advisable to test vitamin B12 levels in all patients with a primary diagnosis of ALS.
3.4.3. Parathyroid hormoneThere are a few reports of patients presenting with weakness and no or minor systemic symptoms or sensory loss with a primary diagnosis of motor neuron disease/ALS subsequently diagnosed with hypercalcemia and hyperparathyroidism (58–62). In some of these studies, no change in disease course was reported after the removal of the parathyroid adenomas, and death subsequently occurred in 3 years. However, significant improvement in neurologic symptoms after resection of the parathyroid adenoma/hyperplasia was reported in others (58). Thereby, it is reasonable to perform serum calcium, phosphorus, and PTH measurement in all patients with a primary diagnosis of ALS.
3.4.4. Paraneoplastic workupAs a paraneoplastic neurologic disorder, MND is a non-classical presentation (63, 64); thereby, by current definition, definite paraneoplastic MND might only be considered in an MND patient when it is associated with cancer and a high-risk antibody. Paraneoplastic MND is very rare, and our knowledge is mostly based on case reports. The most commonly associated antibody is anti-Hu, but occasional reports exist with other antibodies including anti-CV2/CRMP5, anti-Ma2, anti-Yo, and anti-Ri (65, 66). Common underlying malignancies include non-small cell lung cancer, breast cancer, and renal cell carcinoma. There are also reports of associated testicular cancer, ovarian cancer, prostatic carcinoma, and thymoma (65, 67).
Testing for monoclonal gammopathies is controversial in patients with MND. Some studies argue that although monoclonal gammopathies might be more common in ALS patients (68), detecting these gammopathies does not change the management of patients, and immunotherapy or chemotherapy has no effect on the course of MND, hence, no utility in testing for gammopathies (69, 70). On the other hand, other studies suggested that testing might be reasonable since it provides valuable clues for the diagnosis of some common treatable ALS mimics that are associated with gammopathies including motor-predominant multifocal acquired demyelinating sensory and motor neuropathy (MADSAM) and multifocal motor neuropathy. These disorders might be difficult to rule out based on clinical and electrodiagnostic findings (56, 57, 71).
3.4.5. Myasthenia gravis antibodiesThere are several reports of myasthenia gravis masquerading as ALS, especially in muscle-specific kinase (MuSK)-associated myasthenia (72–74). Considering the characteristic bulbar involvement with muscle atrophy in MuSK myasthenia, it can mimic bulbar-onset ALS (74).
In three patients with MuSK myasthenia who had a primary diagnosis of ALS, dropped head syndrome or dysphagia was the early symptom. Weakness occurred more acutely and progressed more rapidly in these patients compared to ALS (72).
3.4.6. Lead levelChronic lead poisoning is another mimicker of motor neuron disease (75, 76). Systemic manifestations, when present, can aid the diagnosis. Neuromuscular manifestations of lead poisoning are varied including polyneuropathy, bibrachial palsy, pure motor neuropathy, and typical motor neuron disease (76).
3.4.7. Hexosaminidase ALate-onset hexosaminidase A deficiency is an uncommon ALS mimic predominantly presenting with lower motor neuron symptoms (77). A PLS-like presentation has also been reported (78).
3.4.8. Recommendations• Neuraxis MRI (brain and spinal cord) should be obtained in all patients diagnosed with ALS to exclude common differential diagnoses (grade B).
• Laboratory screening of the common treatable mimics should be performed in patients diagnosed with ALS (considering age, LMN and/or UMN involvement, and bulbar signs in choosing proper tests). These include complete blood cell count (CBC diff), blood urea nitrogen/creatinine, fasting blood sugar, hemoglobin A1C, thyroid function tests, liver function tests, calcium, phosphorus, PTH, erythrocyte sedimentation rates, serum B12/folate, serum protein electrophoresis/immunofixation, and serum lead level (grade B).
• In patients with bulbar-onset weakness, anti-acetylcholine and anti-MuSK antibodies might be considered (grade B).
• In selected patients, viral markers including HIV, HTLV1, hepatitis B and C, hexosaminidase A in white blood cells or skin fibroblasts, blood very long chain fatty acids, and paraneoplastic panel might be considered (grade B).
3.5. Genetic testingALS has a high estimated mean lifetime heritability of approximately 52.3% (79). Familial ALS, i.e., involvement of at least one first- or second-degree relative, comprises approximately 5–10% of all patients (80). Siblings and children of ALS patients are 10 times more likely to develop ALS (81) with their lifetime risk being approximately 1.4% compared to 0.3% in the general population (79). Most cases of monogenic familial ALS are autosomal-dominant. Currently, more than 30 genes have been found to contribute to ALS risk. Mutations in four major genes including SOD1, C9ORF72, TARDBP, and FUS together account for almost 50% of familial ALS cases, mostly in the form of autosomal-dominant inheritance, and 6% of sporadic cases (82). In a recent Italian study, 27% of ALS patients were carriers of an ALS-related variant, affecting 54.8% of familial ALS, and 17.5% of sporadic ALS patients (83). In Iran and many other Asian countries, SOD1 variants are the most prevalent while C9ORF72 variants are the most common in Caucasian population and Northern European countries (82, 84, 85). Alavi et al. in two consecutive studies sequenced SOD1 and C9ORF72 in 60 and 78 ALS cases, respectively. In the first study, SOD1 variants were found in approximately 12% of the cohort accounting for approximately 40% of the familial cases and 4% of the sporadic cases. In the second study, only 2 of the 78 cases (one familial and one sporadic) had a C9ORF72 variant, revealing a frequency of 6% among familial and 2% among sporadic cases.
The underlying genetic variants can also affect the ALS phenotype. In patients with C9orf72 repeat expansion, the bulbar phenotype is more common while the pure upper motor phenotype is rare. More importantly, C9orf72 variants are the strongest determinant of comorbid frontotemporal dementia (FTD) with ALS (up to 88% of cases). Bulbar presentation is less common with SOD1 mutations, but a flail leg phenotype is more frequent in these patients (86). FUS mutations are the most common genetic variants among patients with juvenile ALS (87). Earlier age of onset and a more aggressive course with normal cognition is more common with SOD1-ALS cases as seen in the p.A4V variant, although other variants such as p.D90A may have longer life expectancy (82, 88). Most TARDBP mutations present with a typical ALS phenotype and no cognitive impairment (82).
Considering the high contribution of genetic variants in both familial and sporadic ALS, it is reasonable to offer genetic testing to those ALS patients who have an affected first- or second-degree relative. On the other hand, uncertainties and complexities in the interpretation of genetic findings, including incomplete penetrance, multiple genetic variants in an individual, poor genotype–phenotype correlation, and pleiotropy, raise important ethical considerations that restrict genetic testing for sporadic cases (89). Nevertheless, genetic testing could be discussed upon the patient's request. One benefit of genetic testing is that it allows patients to be included in related therapeutic clinical trials such as SOD1-ALS (90). In addition, genetic testing is the main diagnostic tool to differentiate some ALS mimics, especially spinobulbar muscular atrophy or Kennedy's Disease, which presents in male patients with slowly progressive pure lower motor neuron syndrome and bulbar involvement. Diagnosis is established by the demonstration of CAG trinucleotide repeat expansion in the androgen receptor gene (91).
3.5.1. Recommendations• Since genetic factors affect the age of onset, progression rate, survival, and disease phenotype of ALS patients, genetic testing should be offered to ALS patients with an affected first- or second-degree relative (grade C).
• For patients with no family history of ALS, genetic testing should not routinely be offered; however, it could be discussed upon patient request (grade B).
• We strongly recommend against genetic testing in asymptomatic individuals (grade A).
3.6. Disease-modifying treatments 3.6.1. Pharmacological agentsRiluzole, a benzothiazole derivative, is the first disease-modifying drug approved by the US Food and Drug Administration (FDA) in 1995 for the treatment of ALS. Multiple clinical trials and systematic reviews showed that riluzole increases overall survival by 3 months and improves bulbar and limb function; however, no significant improvement was found in muscle strength (92). Some studies suggested that increased survival is significantly higher in bulbar-onset compared to limb onset phenotypes (92). The effect of riluzole on the quality of life of ALS patients has not specifically been studied. Riluzole is less beneficial in patients of 75 years or older, those with more severe respiratory involvement (forced vital capacity (FVC) < 60%), or longer disease duration (>5 years of onset), although it is well tolerated in these patients (93, 94). Adverse effects of riluzole include nausea, asthenia, vomiting, diarrhea, anorexia, dizziness, and low hemoglobin (92).
Edaravone, the second drug approved by the US FDA in 2017 for ALS disease modification, is a pyrazolone, with free radical scavenger and neuroprotective properties. The effectiveness of edaravone was shown in a post-hoc analysis (95) performed on the data from the original study (96) and later confirmed in an independent prospective study (97). Therefore, the use of edaravone has been limited to this specific subgroup of patients with (a) age between 20 and 75 years, (b) living independently (grade 1 or 2 in the Japan ALS severity scale), (c) 1–4 scores decrease in ALSFRS-R in the last 12 weeks, (d) score 2 or higher in each item of ALSFRS-R, (e) FVC ≥ 80%, (f) disease duration ≤ 2 years, and (g) probable or definite ALS diagnosis, in addition to absence of any dyspnea, orthopnea, spinal surgery, or renal insufficiency (97). The results from a recent systematic review of three randomized studies on edaravone were indicative of slower disease progression, with 1.63 points on the ALSFRS-R score in edaravone compared to placebo (98). Two recent retrospective large real-world studies showed conflicting results. In the first multicenter study in Germany, the authors reported no difference in disease progression, time to ventilation, or survival between patients receiving edaravone plus riluzole compared to those treated only with riluzole (99). The second study in the US compared the overall survival of ALS patients treated with edaravone to those not treated with edaravone and found a 27% survival benefit (29.5 months vs. 23.5 months) in those treated with edaravone (100).
Edaravone is administered as an intravascular infusion of two 30 mg vials per day for 14 days followed by a drug-free interval of 14 days in the first treatment cycle which is reduced to 10 days in subsequent cycles. An oral suspension has recently been approved by the US FDA. Adverse effects of edaravone are minimal and include headache, bruising, gait disturbance, eczema, respiratory disorder, and glycosuria (101).
On September 2022, the US FDA approved Relyvrio, a combination of sodium phenylbutyrate and taurursodiol for ALS disease modification. It is believed that it causes a reduction in neuronal death via effects on mitochondria and endoplasmic reticulum. In the first phase 2 clinical trial, a slowing of functional decline was observed based on the ALSFR-R score (1.24 per month in the treatment vs. 1.66 in the placebo group) although there was no significant improvement in any of the secondary outcomes including muscle strength, slow vital capacity, hospitalization rate or time to death or tracheostomy (102). ALS patients with less than 18 months of symptom onset under simultaneous treatment with riluzole, edaravone, both, or none were recruited in this study. The treatment regimen consisted of 3g of sodium phenylbutyrate and 1g of taurursodiol once daily for 3 weeks followed by a twice-daily dosing for 21 weeks. The following open-label extension study which assigned all patients to treatment for 35 months showed that the tracheostomy/permanent-assisted ventilation-free survival was significantly longer in patients primarily randomized to the treatment group (25.0 months in the treatment-first vs. 18.5 months in the placebo-first group) (103, 104). Therefore, an early start of Relyvrio increased survival for about 6.5 months.
Following the successful development and US FDA approval of nusinersen, an antisense oligonucleotide for the treatment of spinal muscular atrophy (105), tofersen, an antisense oligonucleotide targeting SOD1gene product, was developed by Ionis/Biogen and tested in a phase III trial on 108 participants with SOD1-ALS (106). Although clinical improvement was not evident after 28 weeks, 12-month data from the open-label extension study showed a slowing in the rate of clinical motor and respiratory decline (107). In addition, neurofilament light, as a marker of disease progression, decreased significantly in cerebrospinal fluid. These findings led to accelerated approval by the US FDA in April 2023. Once available, this drug could be of special interest to Iranian and other Asian ALS patients due to the higher rate of SOD1 mutations in these populations.
3.6.1.1. Recommendations• Treatment with riluzole is strongly recommended for all ALS patients regardless of their disease status (grade A).
• Edaravone should be considered in selected independent ALS patients with 2 years or less of disease duration, mild severity based on ALSFRS-R, and no significant respiratory symptoms (FVC ≥ 80%) (grade A).
• Sodium phenylbutyrate/taurursodiol as a single or add-on therapy is recommended for all ALS patients with less than 18 months of symptom onset (grade B).
• Tofersen as a single or add-on therapy is recommended for all familial SOD1-ALS patients (grade C).
3.6.2. Unapproved treatmentsStem cell therapy is expected to be used for multiple purposes in ALS including preventing further neurodegeneration by producing growth factors, clearance of toxic materials, providing immunomodulatory support, and potential replacement of damaged motor neurons (108). Despite promising results in preclinical studies (109–115), clinical studies are yet inconclusive considering their poor design and small sample size. A recent systematic review and meta-analysis of clinical stem cell studies in ALS found only a transient improvement in ALSFRS-R in studies using intrathecal mesenchymal stromal cells (116, 117). Unexpectedly, this improvement was accompanied by a respiratory worsening based on FVC measurement in these studies. Stem cell therapy in any form has not shown any significant effect on the survival or quality of life of the participants. The major source of bias in these studies was unblinded outcome assessment. Intra-spinal transplantation of neural stem cells has shown a transient improvement in one and no significant effect on disease progression in another study (117, 118). Intrathecal injection of neurotrophic factor-secreting mesenchymal stem cells which showed promising results in a phase 2 study failed to show a significant effect on primary endpoints in the recent phase 3 clinical trial that included 196 ALS patients (119).
3.6.2.1. Recommendations• Stem cell therapy in any form is not recommended in clinical settings (grade C).
• Considering the inconsistent and contradictory results of clinical studies, we also recommend against the use of stem cells in clinical research settings at this time. Further preclinical studies to better characterize the preferred cell type and specifications, dosage, and administration route are needed (grade C).
3.7. Multidisciplinary care managementA multidisciplinary approach is currently considered the standard of care in the management of ALS, which allows providing high-quality care and improving survival and quality of life, while also decreasing complicated hospitalizations and healthcare costs (120–122). However, most patients do not receive timely care in a multidisciplinary ALS clinic. A study in Ireland showed that the time interval between ALS diagnosis to the first visit to a multidisciplinary clinic is approximately 19 months (123). Patients with ALS have several health needs that could best be managed in a multidisciplinary clinic including non-invasive ventilation (NIV), sialorrhea, secretions management, percutaneous endoscopic gastrostomy (PEG) feeding, and behavioral and cognitive disturbances.
While curative treatments are not available for ALS, care in multidisciplinary clinics can increase the survival and quality of life of patients (124). Behavioral and cognitive impairment are important conditions that affect many ALS patients and can disrupt care management in a non-multidisciplinary care setting leading to decreased survival. However, these symptoms can be recognized at earlier stages in a multidisciplinary clinic setting and managed accordingly to decrease their detrimental impact (125). Another important advantage of multidisciplinary ALS care is the initial assessment by a nutrition specialist and the provision of personalized nutritional support that can decrease the rate of severe malnutrition in these patients (126). Rehabilitation is an integral component of a multidisciplinary ALS clinic that can assist patients to continue their independent function safely, enabling them to perform to their fullest potential despite ALS. It is also useful to include a palliative care specialist to manage pain and end-of-life care (127). Social workers have been an important part of the multidisciplinary ALS team in many countries; however, this service is not widely available or covered by insurance in Iran. Further local studies are needed to evaluate the acceptance and role of social workers in the management of ALS patients.
In special situations, multidisciplinary care could be provided via telehealth measures; however, further local studies are needed to evaluate its efficiency, feasibility, and acceptance by patients and providers (128).
3.7.1. Recommendations• All patients must be referred to a multidisciplinary clinic (tertiary center) as soon as an MND diagnosis is made (grade B).
• Multidisciplinary ALS clinics should include neurologists, pulmonologists, speech and language therapists, physical and rehabilitation medicine specialists, nutritionists, gastroenterologists, psychiatrists, and psychologists (grade B).
• All ALS patients should have access to multidisciplinary care. In areas with limited availability of multidisciplinary ALS clinics, a team of local neurologists, pulmonologists, nutritionists, rehabilitation specialists, and psychiatrists must form a local network to manage ALS patients (grade B).
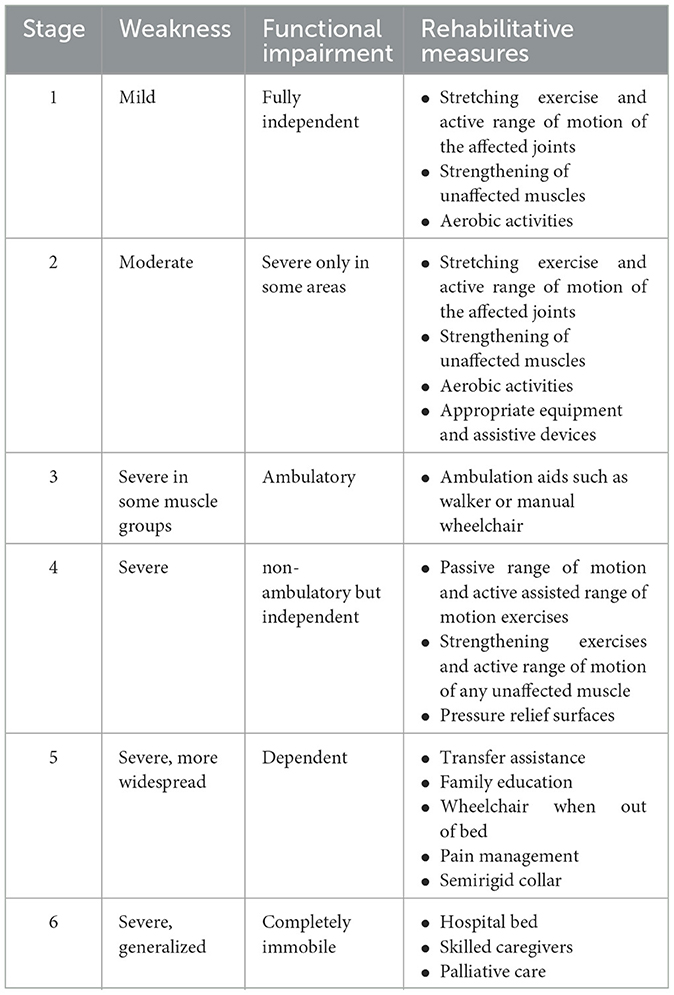
3.8. RehabilitationThe aim of rehabilitation in ALS patients is to improve function, symptoms, quality of life, and survival. Designing a rehabilitation protocol is difficult in patients with ALS because their functional status changes rapidly. Six stages of progression are traditionally being considered in these patients (129). Recommended exercise and rehabilitation measures for each stage are shown in Table 2.
Table 2. Rehabilitative measures based on Sinaki stages.
The most widely used rehabilitation modality in ALS is exercise. Exercise has traditionally been avoided in ALS patients believing it might worsen the course of the illness. More recent studies including recent systematic reviews and meta-analyses have shown that range of motion and stretching exercise is safe and effective in these patients which led to the inclusion of exercise as part of standard ALS care (130–132). Exercise may help to maintain function and prevent contracture and pain and, in the long term, can improve functional ability and pulmonary capacity. Endurance/aerobic exercise seems to be superior to resistance exercise in ALS, and moderate-intensity exercise is preferred over high-intensity exercise (131). Patients should be encouraged to start daily exercise early in the disease course. The intensity of exercise should be modified such that to avoid fatigue, dyspnea, or cramp which are signs of overuse weakness. Pain and fatigue that continue longer than 30 min after exercise are other indicators that show the exercise program must be modified (133). On the other hand, prolonged interruption of rehabilitation may accelerate functional motor decline in ALS patients (134).
Assistive devices and orthoses such as ankle foot orthosis, night splint, wrist extension orthosis, thumb positioning orthosis, and cervical collar, as well as adaptive equipment and assistive devices including cane, walker, and wheelchair, are used to assist in the improvement of function and mobility in ALS patients.
Evidence is low or lacking about other modalities such as breathing exercises, resistance exercises, aqua therapy, electrical stimulation, and ultrasound (131).
3.8.1. Effect of rehabilitation on pain, spasticity, and fatigueContracture and immobilization are important contributors to pain among ALS patients. Therefore, night-splints, shoulder approximation sleeves, and dynamic splints have been proposed to help keep ankles and hands in a neutral position, prevent shoulder subluxation, and reduce the risk of contractures. Regular skin check is necessary to prevent complications including pressure sores or pain. Proper lumbar support for wheelchair-bound patients may reduce low back pain. Gait training, appropriate transfer techniques, and modification of home and workplace to reduce the risk of falls and injuries may help in maintaining function and preventing pain (135–138). Physical modalities including ice and heat may be used to reduce pain.
In the management of spasticity, rehabilitation measures are the most important options. Stretching and a range of motion exercises, cold pack, transcutaneous electrical nerve stimulation, shock wave and ultrasound, night-time neutral position splinting, and hydrotherapy are suggested; however, more studies are needed (139, 140).
Exercise, work simplification, and energy conservation techniques including pacing and regular rest periods between activities may be of assistance in reducing fatigue in ALS patients. Using proper orthosis with light weight material and wheeled walkers instead of standard walkers is also recommended. When appropriate, manual or powered wheelchairs or scooters can be used to reduce fatigue. A custom-fitted wheelchair is a better choice to support the spine and provide pressure relief.
3.8.2. Recommendations• Range of motion and stretching exercises are effective and recommended in all ALS patients (grade B).
• Ambulatory patients might benefit from aerobic and endurance exercise (grade A).
• Moderate-intensity strengthening exercises should be reserved for patients with high functional status (grade A).
• Exercise should not cause pain or fatigue that lasts more than 30 min, overuse weakness, cramps, or dyspnea (grade E).
• Proper use of splints and sleeves reduces the risk of painful contracture and subluxation (grade E).
• Gait training programs and home and workplace modifications are recommended to prevent falls and injuries (grade E).
• Patients' and caregivers' education should include appropriate transfer techniques and proper wheelchair back support (grade E).
• Use of orthoses and assistive and adaptive devices should be considered on an individual basis (grade E).
• Range of motion exercise and stretching as well as proper orthosis or physical modalities are useful to decrease spasticity (grade E).
• Interventions to reduce fatigue include energy conservation techniques, change to lightweight braces, proper assistive devices and mobility aids, and exercise programs including stretching, endurance, and aerobic exercise (grade E).
3.9. Management of swallowing, communication, and nutrition 3.9.1. Speech therapy evaluations and interventionsSpeech motor problems, although more severe and frequent in bulbar-onset form, have a lifelong prevalence of over 80% in all ALS patients which lead to complete loss of oral communication skills in 75–95% of patients. All subsystems are involved in ALS, especially respiration, phonation, articulation, and resonance. Bulbar weakness leads to anarthria after about 18 months (141). Cognitive dysfunction, especially involving executive, social, and language domains, also accounts for loss of communication abilities in ALS patients.
Dysarthria in ALS is typically of mixed type with varying combinations of flaccid and spastic features, usually starting with nasality leading to a decrease in speech rate and loss of intelligibility in later stages. More severe bulbar weakness might lead to hypernasality and a change in voice quality; nevertheless, phonation is preserved even in later-stage ALS (142).
High-quality studies are lacking about the management of dysarthria and dysphagia in ALS despite their universal presence in ALS patients. In the early stages, training patients to speak more slowly could be helpful to increase intelligibility and adapt to respiratory weakness. Strengthening exercise of the bulbar muscles and diaphragm is controversial; however, the general exercise rules stated in previous sections could reasonably be applied here (143–145). Palatal lift has been suggested for patients with significant dysarthria due to hypernasality. In advanced stages, many patients with ALS will need to use augmentative assistive communication technologies. There is a notably high acceptance rate of these technologies among ALS patients and reportedly 46% of patients continue to use the technology during their last week of life (13–15).
Dysphagia, mostly of oropharyngeal type, is an early presentation in 20–30% of the patients. In later stages, nearly all are affected which leads to decreased swallowing safety and efficacy (146). Patients with mild-to-moderate dysphagia might benefit from swallowing aid techniques. Recently, the “Ishizaki Press Method” has been shown to be effective for dysphagia in a patient with severe ALS (147). This method involves the application of finger pressure on specific maxillofacial points and is believed to play a role in triggering the swallowing reflex.
3.9.1.1. Recommendations• It is recommended that all patients with ALS be evaluated by an expert speech and language pathologist at the first signs of disease and treated and followed up every 3 months or as indicated in speech therapy service during the mild-to-moderate stages (grade B).
• Dysphagia intervention should be considered according to the disease stage:
a. In the early stages of ALS with normal swallowing, speech pathologist consultation should be offered (grade B).
b. In the presence of occasional problems with eating and drinking, interventions include modification strategies, postural adjustments, and swallowing maneuvers (grade B).
c. When encountering moderate problems with eating and drinking, dietary consistency changes are recommended (grade A).
d. In the more severe stage, feeding tube placement will be a safe option. High-viscosity liquids should be handled with caution (grade B).
e. For all stages, with or without eating ability, oral hygiene should be practiced throughout the day (grade A).
• For dysarthria, using slow speech rate, exaggeration of articulation, improvement of respiratory efficiency through phrasing, tongue strengthening exercises, and diaphragmatic exercises are recommended (grade B).
• At any stage when a patient with ALS cannot communicate the use of augmentative assistive communication strategies should be considered to enhance the quality of life (grade B).
3.9.2. SialorrheaDrooling or sialorrhea is not only a troubling symptom in patients with ALS but also increases the risk of aspiration pneumonia. The prevalence of sialorrhea in ALS patients is approximately 20–50% (148). Normal daily production of saliva is about 1.5 liters, and its clearance depends on the function and strength of the bulbar, facial, and buccal muscles. Dysphagia and weakness of these muscles due to ALS lead to the pooling of saliva and eventually drooling.
Excess salivation directly or indirectly influences ALSFRS-R scores, mostly in the first three items (speech, salivation, and swallowing); therefore, the overall function and quality of life will improve with the successful management of sialorrhea (149). Moreover, in ALS patients who are using NIV, the pooling of oral secretions may lead to decreased tolerance and efficiency of NIV, the consequences of which are hypoxia and aspiration pneumonia. It has been shown that patients with normal oral secretion scores better tolerate NIV and have longer survival compared to those with severe sialorrhea (150). Physicians should be aware that although sialorrhea is troublesome, care should be taken to avoid overtreatment leading to dry mouth since it reduces dental hygiene and aggravates swallowing difficulties.
Different options for the management of sialorrhea include behavioral techniques (e.g., deliberate swallowing), frequent suction, anticholinergic agents, botulinum toxin injection, and radiotherapy. Anticholinergic agents are the most common first-line treatment used in the management of sialorrhea. In one study, 61% of patients with sialorrhea showed some degree of improvement (151). However, side effects especially in elderly patients remain a major concern (152).
The second therapeutic option for the management of sialorrhea resistant to anticholinergics or when side effects develop is local botulinum neurotoxin injection (153). Its efficacy has been approved in a number of studies with effects lasting up to 4 months and side effects being negligible (154). There is generally no significant difference between botulinum toxins type A and B (155).
Radiation to the salivary glands is another therapeutic option for the management of sialorrhea that should be reserved for patients who are refractory to other options (136, 156). The total dose of 20Gy divided over 4–5 fractions seems to be the most effective; however, tolerability might be an issue in some patients in whom lower doses could be tried (e.g., 8 Gy in one fraction) (120, 157).
3.9.2.1. Recommendations• Sialorrhea should be actively evaluated and appropriately managed in ALS patients to improve quality of life and survival (grade A).
• In the management of sialorrhea, it is important to avoid dry mouth which may lead to poor dental/oral health and dysphagia (grade A).
• Anticholinergic agents are recommended as the first-line therapeutic option (grade A).
• In cases of anticholinergic resistance or significant side effects, botulinum neurotoxin injection into the salivary glands should be considered (grade B).
• Salivary gland radiation should be reserved for patients who are refractory to anticholinergic agents and botulinum neurotoxin injection (grade B).
3.9.3. Nutrition interventionsNutritional status has an important role in determining the prognosis among ALS patients (158), but unfortunately, malnutrition affects 16–55% of ALS patients (159) which together with progressive muscle wasting leads to significant weight loss. Therefore, regular nutritional assessment at diagnosis and every 3 months is recommended by all available guidelines (120, 136, 153). The assessment includes a history of recent weight loss, current food intake, and dysphagia assessment as well as lipid profile, nutrient levels, and evaluation of nutrition-related complications. Although weight and body mass index (BMI) measurements are helpful in detecting malnutrition, body composition analysis using dual-energy X-ray absorptiometry (DEXA) or bioelectrical impedance analysis provides valuable information for distinguishing between loss of fat tissue versus muscle atrophy. More frequent monitoring might be necessary in patients with known malnutrition or dysphagia (160). Any indication of weight loss or malnutrition should be treated aggressively as soon as diagnosed considering the grave consequences. Food fortification is usually the first step due to its convenience and fewer complications (160). Measurement of energy expenditure in ALS patients can inform the decision to offer various nutritional interventions. Indirect calorimetry provides a more reliable measurement of energy expenditure; however, if not available, this could be estimated via the Harris–Benedict equation using the patient's nutritional status, malnutrition history, physical activity, and ventilation condition (161). Generally, non-ventilated patients have higher energy requirements.
A water swallow test or volume viscosity swallow test could be used for nutritional evaluation of dysphagia. In ALS patients, dysphagia is usually more prominent with thin liquids; therefore, food with soft, semisolid, or semiliquid consistency is preferred (153, 160, 162). In moderate dysphagia, dietary counseling may include texture modification as well as patient education to perform postural maneuvers, chin tuck, head rotation, and throat clearing (160, 163). Home parenteral nutrition is generally not indicated in ALS patients (153, 160, 164).
3.9.3.1. Recommendations• Regular nutritional assessment by a nutritionist including nutritional history, clinical examination with swallowing evaluation, and weight and BMI measurement should be performed at baseline and at least every 3 months in all patients (grade A).
• Objective measurement of body composition and energy expenditure might be considered on an individual basis (grade A).
• Nutritional counseling includes a discussion of food fortification, oral nutritional supplementation, and the possible need for early enteral nutrition such as PEG. Food enrichment is recommended when weight loss, fatigue, or effortful feeding presents, while oral nutritional supplementation is recommended for patients with unmet energy and nutrient requirements (grade A).
3.9.4. Gastrostomy feedingConsidering the high frequency of swallowing impairment in ALS, evaluation of swallowing function should be performed early in the disease course in all patients. Patients with signs of swallowing impairment will benefit from a more comprehensive instrumental swallowing evaluation, including a video fluoroscopic swallow study (VFSS) or fiberoptic endoscopic evaluation of swallowing (FEES). During clinic visits, assessment of swallowing function may include eating and swallowing questionnaires, dietary intake, examination of bulbar function and dysphagia, pulmonary function and airway clearance, and estimation of aspiration risk (165). Swallowing function could also be estimated based on the ALSFRS-R, pulmonary function tests, and regular body weight measurements (166, 167). Eating assessment tool-10 (EAT-10) is a validated measure to detect unsafe swallowing. Patients with EAT-10 scores of 8 or higher are at three times greater risk of aspiration (168). Previous guidelines introduced weight loss of at least 10% relative to baseline as an indication for PEG placement in ALS patients (136). Recent large prospective studies confirmed this indication and showed that patients with weight loss of <10% had better survival after PEG placement (169). Similarly, a BMI of <18.5 is a marker of undernutrition and an important nutritional survival marker for ALS which serves as a guide for nutritional interventions (170). Another important consideration in the decision for PEG placement is the respiratory function status. Decreased FVC of 50% or less almost precludes PEG placement because of the intolerance to the required sedation at this stage. A large retrospective study combined with a meta-analysis of the results of all previous studies showed that a significant increase in survival follows PEG placement, especially when FVC is 50% or more than predicted at the time of the procedure (171). Another meta-analysis compared the efficacy and safety of PEG vs. nasogastric tube (NGT) in patients with various causes of swallowing impairment and found that intervention failure occurred less with PEG compared to NGT; however, there was no significant difference between the groups regarding rates of mortality, adverse events, weight change, pain, or ease of learning (172). Expectedly, compared to NGT, PEG was more convenient for patients with less interference with social activities. Therefore, considering the clear benefits, NGT could be considered if PEG is not practicable.
3.9.4.1. Recommendations• Enteral tube feeding should be considered if there is a failure of management by speech therapy or diet modification measures with weight loss approaching 10% of baseline, BMI < 18.5, unsafe swallowing in instrumental tests, or if FVC is approaching 50% irrespective of the degree of swallowing impairment (grade A).
• PEG and NGT have similar effectiveness in maintaining good dietary intake, but regarding quality-of-life measures, PEG is superior (grade A).
3.10. Assessment and management of respiratory failureAn updated Cochrane review showed that NIV improves the quality of life and prolongs survival of ALS patients with a normal to moderately impaired bulbar function by 205 days, although it did not improve survival in those with severe bulbar weakness (173). Therefore, it is important to determine the need and timing of NIV in ALS patients. After the initial ALS diagnosis, the indicators of respiratory failure should carefully be examined, although many patients might not manifest respiratory symptoms. FVC measurement is traditionally considered the gold standard of respiratory assessment; however, due to the high frequency of bulbar and lip weakness in ALS patients, a standard spirometry cannot be performed in some patients or might generate inaccurate information. In fact, some studies propose that other tests such as slow vital capacity (SVC), supine FVC, maximal inspiratory (MIP) and expiratory pressures (MEP), peak cough flow (PCF), and sniff nasal-inspiratory pressure (SNIP) are more accurate in these patients for the evaluation of respiratory function and decision about NIV initiation (174–177). Maximum cough expiratory flow demonstrates the effectiveness of cough and airway clearance. PCF can be considered as an indicator of expiratory muscle function which measures how voluntary cough can affect the risk of aspiration pneumonia.
Some ALS patients may have normal FVC measurements despite nocturnal desaturation. Nocturnal desaturation indicates weakness of the respiratory muscles and can be considered an indication for the use of NIV. Therefore, more frequent nocturnal oximetry is advisable in the respiratory assessment of ALS patients. ALS patients usually have a progressive decline in sleep quality which affects the quality of life and survival of these patients. Considering the limitations of performing a standard polysomnography in ALS patients, sleep apnea could be assessed by a combination of oximetry or home respiratory polygraphy plus transcutaneous or end-tidal carbon dioxide monitoring (178, 179).
The first evaluation of respiratory function usually consists of clinical history and examination aiming to reveal any tachypnea, dyspnea, orthopnea, paradoxical respiration or use of accessory muscles, nocturnal desaturation, and daytime headache or sleepiness. The Epworth sleepiness scale could be used for the evaluation of daytime sleepiness (scores above 9). Any sign of respiration impairment should then trigger further evaluation using one of the abovementioned modalities. Usually in the presence of one of the findings such as FVC < 80%, SNIP < 40 cmH2O, pCO2 > 45 mmHg, or nocturnal desaturation, the use of NIV is on the agenda (136). FVC decreases more slowly in patients who start NIV earlier (180). In fact, more recent studies have shown that even earlier initiation of NIV, with FVC ≥ 80%, further increases survival in ALS patients (181, 182). Factors such as the use of cough aids, control of secretions, and proper nutrition can improve the effectiveness of NIV.
Respiratory infections are frequent in ALS patients due to inactivity, sialorrhea, cough inefficiency, and dysphagia. In such cases, the risk of developing pneumonia can be minimized by methods such as vaccination against influenza and pneumococcus. Cough is the main airway protection mechanism, but it is considerably weak in many ALS patients. Assisted cough devices are useful when peak cough flow falls below 270–300 L/min (183). Existing evidence confirms the beneficial role of these physiotherapy interventions in improving respiratory complications and increasing the survival of ALS patients (184). Studies show that mechanical insufflation/exsufflation and the breath-stacking technique are associated with a reduction in adverse respiratory complications (185, 186).
3.10.1. Recommendations• Signs of respiratory impairment such as daytime headache, sleep disturbances, weak cough, and labored breathing should be examined in all ALS patients on every visit and at least one or two times every 3 months (grade A).
• FVC monitoring, SNIP, maximum inspiratory/expiratory pressure, or supine FVC testing (in patients with normal upright respiratory tests) must be done in all patients at least once every 3 months (grade A).
• NIV should be initiated in patients with FVC < 80%, SNIP < 40 cmH2O, pCO2 > 45mmHg, significant desaturation on overnight oximetry, or when signs/symptoms of respiratory weakness are present (grade A).
• Patients and caregivers should receive education about breath-stacking techniques.
• Patients with decreased cough or PCF of less than 270 L/min in whom medical treatment has failed to manage secretions or is not an option, assisted cough devices should be offered (grade B).
3.11. Symptom management 3.11.1. PainPain is extremely common during the course of ALS which sometimes might start even before the onset of motor dysfunction (187). Depression and reduced quality of life have a significant association with pain in ALS patients. The first step in pain management is proactive screening which means that the patients' self-report is insufficient, and physicians should actively inquire about pain (153, 188). The second step is to identify the source. Nociceptive pain due to secondary causes (e.g., joint deformities and skin pressure) is the most common type of pain especially as disability increases. Nociceptive pain responds well to non-steroidal anti-inflammatory drugs and paracetamol which make the first line of treatment in these patients (189–191). Nociceptive joint pain can also be treated with local injection of steroids or lidocaine (188, 192). Gabapentin, pregabalin, and tricyclic antidepressants are recommended when pain has a primarily neuropathic nature (120, 188, 193). In a more advanced stage of the disease, opioids are reported to be effective (190, 191). Cramps and spasticity are other major causes of primary pain in ALS and should be managed accordingly (188, 194).
3.11.1.1. Recommendations• In the treatment of nociceptive pain, non-steroidal anti-inflammatory drugs and paracetamol should be considered in the first line. As the second line, intra-articular steroids or lidocaine injection might be considered (grade B).
• Gabapentin, pregabalin, and tricyclic antidepressants could be considered in patients with a neuropathic type of pain (grade B).
• In selected patients with advanced di
留言 (0)