
記住我
Coronary artery abnormalities include abnormal number and origin, while single coronary artery (SCA) is relatively rare, accounting for 0.031% in coronary angiography (1, 2) and 0.024%–0.066% in the general population (3, 4). It is unclear whether SCA is an isolated congenital heart disease or is associated with other congenital abnormalities. Dilated cardiomyopathy (DCM) is currently defined by the presence of left ventricular or biventricular dilatation and systolic dysfunction in the absence of abnormal loading conditions (hypertension, valve disease) or coronary artery disease sufficient to cause global systolic impairment (5, 6). The causes of DCM are heterogeneous (7), and we believe this is the result of genetic predisposition interacting with extrinsic or environmental factors (8, 9). The SCN5A gene mutation is associated with a range of clinical diseases. Here, we present a rare case of SCA with DCM accompanied by the SCN5A gene mutation.
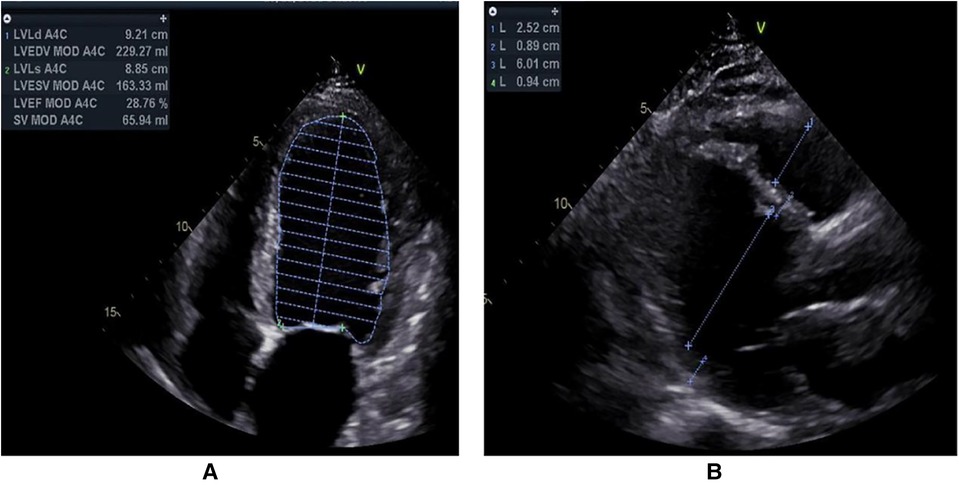
2. Case presentationA 55-year-old male was admitted to our hospital because of dyspnoea. That day, he felt dyspnoea after 200 metres of flat walking. Emergency medical services were called, and he was transported to the emergency department at our hospital. On evaluation, the systolic blood pressure was 116/91 mmHg, the pulse was 115 beats per minute, the respiratory rate was 17 breaths per minute, and the oxygen saturation was 100%. An electrocardiogram (ECG) showed sinus bradycardia with T-wave inversions and premature ventricular contractions (Supplementary Figure S1). On arrival, the patient reported paroxysmal dyspnoea. He had multiple similar episodes during the previous 20 days, without fever, cough, vomiting or diarrhoea. The symptoms were usually provoked by physical exertion, mental stress or intense emotion. Evaluations at other hospitals showed chronic bronchitis and emphysema, an enlarged left ventricle and decreased cardiac function, and further treatment was recommended. The patient had no other illnesses. His family history was unremarkable. He was a farmer. He drank alcohol occasionally, and had smoked in the past 30 years, and did not use illicit drugs or herbal preparations. There were no recent exposures to ill persons. Physical examination showed oedema of both lower limbs, while the other limbs were normal. Levels of sodium, chloride, carbon dioxide, D-dimer, magnesium and tests of liver function and renal function were normal. Troponin I and serum NT-proBNP were rising. A 24-h Holter monitor revealed occasional atrial premature beats, frequent multiple ventricular premature beats and ST-T changes. Transthoracic echocardiography (TTE) showed decreased systolic function (LVEF = 29%), an enlarged left ventricle (LVEDD = 60 mm), cardiomyopathy, moderate mitral regurgitation and moderate pulmonary hypertension (Figure 1). There was no family history of cardiovascular disease. Drugs improving microcirculation and cardiac function were administered, and he was then admitted to our ward. Laboratory tests were conducted, and the results are shown in Table 1.
Figure 1. Transthoracic echocardiography was routinely examined from left parasternal long-axis view (A) and apical four-chamber view (B).
Table 1. Laboratory data.
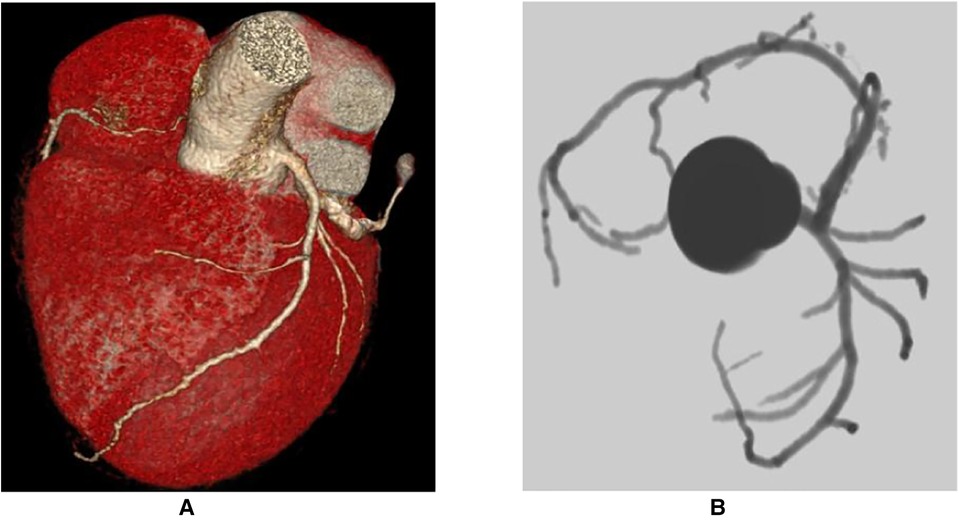
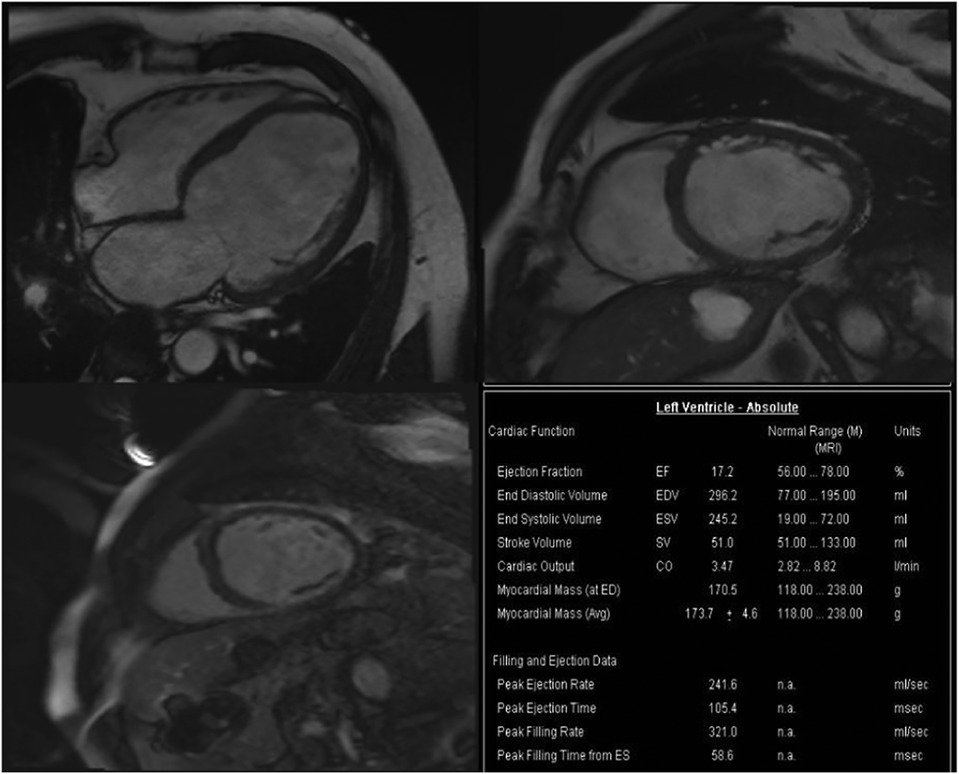
Based on the above clinical examination, computed tomography coronary angiogram (CTCA) was performed for the patient to rule out atherosclerotic coronary artery disease. To our surprise, the patient had a very rare SCA, a congenital abnormality of the coronary artery system that may provide low perfusion to the entire heart muscle, which causes chest pain, angina or dyspnoea. CTCA showed that a coronary artery from Valsalva's left sinus was divided into the left anterior descending branch (LAD) and the left circumflex branch (LCX). The distal end of the LCX continued its course beyond the crux into the atrioventricular groove, supplying the right atrium and right ventricle with a superdominant LCX without stenosis and a calcium score of zero Agatston units (Figure 2). Because no intervention would be appropriate in the absence of significant coronary artery stenosis in CTCA, we decided not to perform invasive coronary angiography. To identify the cause of heart failure, we performed cardiovascular magnetic resonance (CMR) on the patient. CMR showed reduced left ventricular systolic function (LVEF = 17.2%), left ventricular enlargement (LVEDV = 296.2 ml), thinning of the myocardium and abnormal delayed reinforcement in the basal segment of the ventricular septum, which was consistent with the diagnosis of DCM (Figure 3). Although improved cardiac imaging techniques have made endomyocardial biopsy (EMB) less necessary, it has traditionally been used to confirm the aetiology in some forms of DCM. However, EMB is no longer frequently performed and was not conducted in this case.
Figure 2. Computed tomography coronary angiogram. (A) Course of the LCx on the posterior atrioventricular groove and continuation of its course in the RCA territory along with take-off of the posterior descending artery. (B) Absence of take-off of the RCA from the right coronary sinus of Valsalva (yellow arrow) and normal origin of the LMS which bifurcates into the LAD and LCX. LAD, left anterior descending artery; LCX, left circumflex artery; LMS, left main stem artery; RCA, right coronary artery.
Figure 3. Cardiac magnetic resonance imaging showed DCM. CMR detected dilation of the left ventricle (LVEDV = 296.2 ml), reduction of cardiac systolic function (CO = 3.47l/min, LVEF = 17.2%) and increase of left ventricular mass (LVM = 170.5 g), no signs of storage disease or inflammation. CMR, Cardiac magnetic resonance imaging; LVEDV, left ventricular end-diastolic volume; LVEF, left ventricular ejection fraction; CO, cardiac output.
We suspected that the mutation of a certain gene caused the patient to have SCA and DCM, therefore, we arranged a clinical gene test of the whole exon. Genetic screening showed the SCN5A; NM_198056.2:c.1858C > T (p. Arg620Cys) mutation (10–13) and the pathogenicity of this variant has been reported; SCN5A; NM_ 198056.2:c.1008 g > a [P. (pro336=)] mutation and there is no report on the pathogenicity of this variant. Sanger sequencing result is shown in Figure 4 and NSG data is shown in Supplementary Table S1. The SCN5A C.1858C > T (P. arg620Cys) mutation may be associated with DCM in this patient. At the same time, APOA5; NM_052968.4:c.990_993delAACA (p. Asp332Valfs*5) was mutated in this patient, and its pathogenicity has been reported. According to ACMG Guidelines, this variant is considered a suspected pathogenic variant (14). The APOA5 gene mutation results in hypertriglyceridemia or hyperlipoproteinemia type 5, which was in line with the diagnosis of hypertriglyceridemia based on the patient's blood test. The relationship between SCA and DCM was not elucidated by genetic testing. There was no previous report on the absence of a RCA with DCM. According to findings of CTCA, TTE, CMR and other test results, we considered that the occurrence of DCM in this patient may not due to the absence of the RCA. Rather, we supposed that a gene mutation may cause the SCA and DCM.
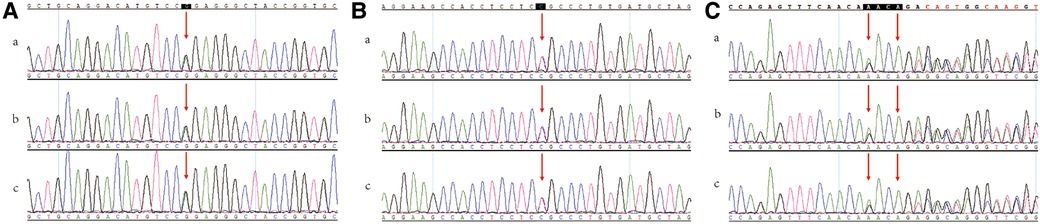
Figure 4. Sanger sequencing data. (A) SCN5A; NM_198056.2; c.1858C > T; p.Arg620Cys. (B) SCN5A; NM_198056.2; c.1008G > A; p.(Pro336=). (C) APOA5; NM_052968.4; c.990_993delAACA; p.Asp332Valfs*5. (a) proband (b) the son of proband (c) the daughter of proband.
Therefore, we performed family verification on the two children of the patient. The children of the proband both carried the SCN5A: c.1858C > T(p. Arg620Cys)/c.1008G > A [p.(Pro336=)] variant and APOA5: c.990_993delAACA (p. Asp332Valfs*5) variant.
3. DiscussionThis is a rare case of congenital absence of the RCA in a patient with a DCM-related gene mutation and hyperlipidemia. Our patient had tested positive for a gene variant known to cause DCM and had 2 months of chest tightness and dyspnoea after activity along with hyperlipidemia. The son and daughter of the proband also tested positive for the same mutation. We did not perform a full pedigree of the family since other relatives refused to be tested for the relevant mutations.
3.1. Genetic DCMThe prevalence of DCM and of genetically mediated DCM is not fully known because of geographic variations, patient selection and changes in the diagnostic criteria (15–18). The European Society of Cardiology Working Group on Myocardial and Pericardial Diseases presented an update of the existing classification scheme of cardiomyopathies in 2008. They grouped the cardiomyopathies into specific morphological and functional phenotypes, including hypertrophic cardiomyopathies, DCM, arrhythmogenic right ventricular cardiomyopathies (ARVC), restrictive cardiomyopathies and unclassified cardiomyopathies. Each phenotype was then subclassified into genetic and nongenetic forms, but overlaps exist between the two groups (6, 8). Meanwhile, studies have shown that 20%–50% of non-ischemia cardiomyopathy patients have a family history (19). All of these findings suggest that genetic factors play an important role in the pathogenesis of cardiomyopathy.
Familial patterns or genetic causes have been identified in up to 35% of cases of idiopathic DCM (20). The genetic causes of DCM are diverse. An autosomal dominant trait is the most common, while autosomal recessive, X-linked and mitochondrial inheritance patterns are less common (21). TTN and LMNA are the main genes associated with the predominant cardiac phenotype, accounting for up to 25% and 5% of all cases of autosomal dominant DCM, respectively (22). RBM20 and DES are also common in clinical practice (23, 24). Other genetic causes of a predominant DCM phenotype include mutations in sarcomere genes, Z-disc protein-encoding genes, genes encoding desmosomal proteins and genes associated with ion channel function (10–29).
The SCN5A gene is associated with ion channel function caused by DCM. With the development of gene detection technology, an increasing number of individual genes have been associated with inheritance in familial DCM cases. Cheng Shen et al. performed a cross-sectional study in Chinese patients with sporadic DCM and suggested that MYBPC3, SCN5A, MYH7, MYPN and LDB3 are the major genes hosting the at-risk genomic variants of sporadic DCM (30).
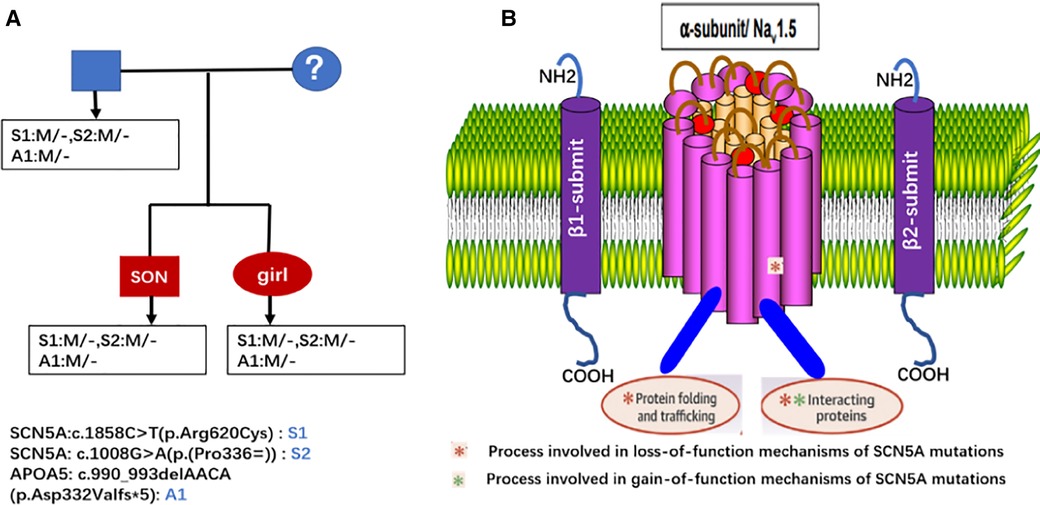
3.2. SCN5A gene mutation-related clinical diseasesThe SCN5A gene, located on chromosome 3p21 with 28 exons, encodes the alpha subunit of the main sodium channel Nav1.5, which enables the rapid influx of Na+ ions (INa) (19). This alpha subunit is expressed in human cardiomyocytes as well as in many other tissues and cells, such as embryonic and denervated skeletal muscle cells in the brain, interstitial cells of Cajal in the human jejunum and colon and smooth muscle cells (31–36). Some studies have shown that the SCN5A splice variant is also expressed in macrophages, where it activates innate immune signalling for antiviral defence (37–39). Voltage-gated Na+ channels are crucial in the excitation and propagation of electrical impulses in cardiomyocytes (40). Nav1.5 interacts with several proteins, including ancillary β-subunits, fascia adherens junctions, desmosomes, gap junctions and intracellular proteins that regulate the gain-of-function and loss-of-function of Nav1.5 proteins (Figure 5) (41). SCN5A gene mutations are associated with a clinical spectrum, including Brugada syndrome, long-QT syndrome, progressive cardiac conduction disease, sinus node dysfunction, atrial fibrillation, DCM, multifocal ectopic Purkinje-related premature contractions, irritable bowel syndrome (IBS) and other gastrointestinal disorders, such as chronic idiopathic intestinal pseudo-obstruction (32, 34, 41, 42).
Figure 5. Genetic test results of the proband and his children, both the proband and his children had the same three genes mutation and were heterozygous. (A) Molecular structure of the SCN5A translated into the a-subunit (Nav1.5) of the cardiac sodium channel. (B) Nav1.5 consists of an N-terminus, C-terminus and 4 structurally homologous domains.
3.3. SCN5A gene mutation and DCMThe genetic background of patients with DCM is complex: 7% have a single heterozygous mutation, more than 38% have a compound heterozygous or combined mutation, and 12.8% have three or more mutations (43–45). Although many previous works strongly implied that the SCN5A gene mutation plays a critical role in the development of cardiomyopathy, the mechanisms remain controversial. Several mechanisms have been postulated. SCN5A gene mutations related to tachyarrhythmia or other conduction abnormalities induce DCM, especially for some patients with a long history of arrhythmia (41, 46–49). A primary disruption of Nav1.5 can affect cellular pH and Ca2+ homeostasis and result in a DCM phenotype (42, 50–52). The SCN5A channel mutation disrupts the sodium channel domain to target the appropriate cytoskeletal components, such as syntrophins and dystrophins (48, 53–57). In addition, the environment and common gene variants may act together with SCN5A mutations to cause DCM (42, 52, 58, 59).
The proband and offspring carried the same three mutations, but they have not shown signs of DCM, and we will continue to follow the family.
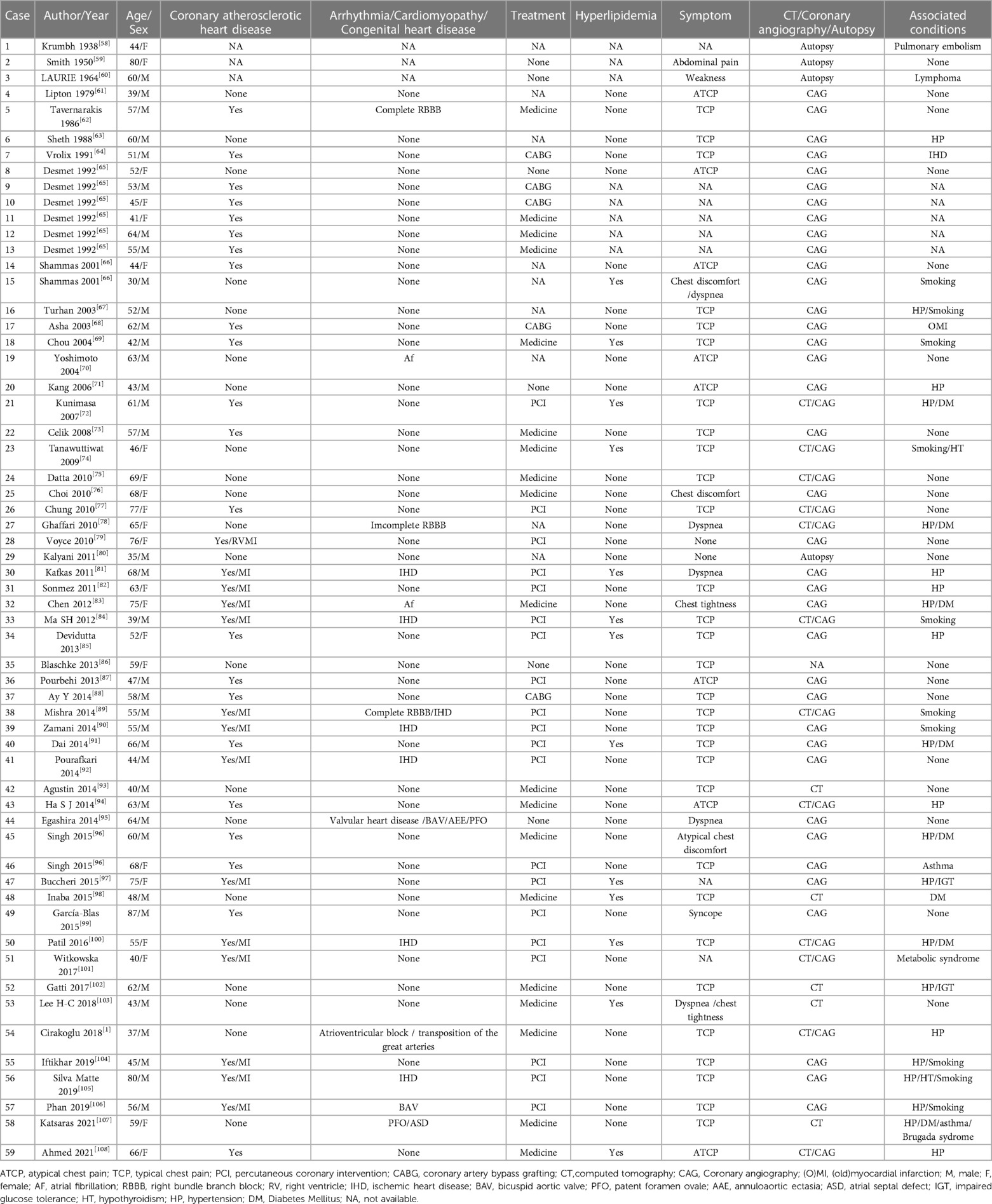
3.4. SCA: absence of RCAIn 1979, Lipton et al. defined and classified SCA, which can be divided into 9 types according to origin and anatomical process. The cause of SCA is still unknown (60). Although isolated reports indicate that specific coronary abnormalities occur in families, no clear pattern of coronary inheritance has been found in humans (61). We reported a rare L-I pattern of a SCA according to the Lipton classification and summarized a review of identical types of SCA literature, in which the RCA was absent and the LCX was markedly dominant and nourished the right ventricle and atrium beyond the atrioventricular groove. A careful review of the literature revealed 59 cases with a similar anomalous coronary origin and pattern, including demographic characteristics, symptoms, complications, diagnosis and treatment strategy (Table 2) (62–112). Of these 59 patients, 22 were male and 37 were female, and their mean age was 56 years (range 30–87 years).
Table 2. Summary characteristics of L-1 of SCA.
3.4.1. SCA and coronary heart diseaseSCA is generally considered to be a benign abnormality; however, some authors have reported that 15% of SCA patients develop myocardial ischaemia as a direct consequence of coronary artery abnormalities (78). SCA abnormalities showed a higher risk of coronary atherosclerosis in a study (113). However, the relationship between congenital abnormalities and atherosclerosis is controversial. Tanriverdi H and Rudan D suggested that atherosclerosis in patients with coronary artery abnormalities was a coincidence (114, 115). According to Shirani's review, 15% of patients with isolated SCA have evidence of myocardial ischaemia without significant atherosclerotic stenosis (116). There were 36 (61%) patients with coronary artery absence combined with coronary atherosclerotic heart disease (CHD); of them, coronary artery bypass grafting was performed in 5 patients, and percutaneous coronary intervention was conducted in 20 patients, which was higher than the general population. SCA anomaly by itself is unlikely to induce myocardial ischaemia, and it has been considered a benign lesion (98). We consider that SCA promotes the occurrence of CHD. When evaluating clinical symptoms and the degree of myocardial area at risk, it is extremely crucial to refer patients with combined SCA and CHD for selective coronary revascularization. Previous studies have shown that interventional surgery for SCA with atherosclerosis has potentially serious consequences; thus, it is rarely performed (92, 102). However, our report revealed that the proportion of patients undergoing interventional surgery in this condition was as high as 69% (25/36). Therefore, interventional operation can be performed successfully in a patient who have SCA with CHD when the anatomy is appropriate.
3.4.2. SCA with congenital heart diseaseCongenital heart disease is uncommon in the L-I pattern of SCA, accounting for 7% (4/59). Patent foramen ovale and bicuspid aortic valve were reported in 2 patients, and one of them had annuloaortic ectasia leading to heart failure (101, 105, 107). Congenitally corrected transposition in a situs inversus was shown in a patient (1).
3.4.3. SCA with arrhythmiaOverall, 10% (6/59) of SCA patients have shown arrhythmia, including 3 patients with right bundle branch block (68, 85, 102), 2 patients with atrial fibrillation (30, 43), 1 patient with atrioventricular block (72) and 1 patient with Brugada syndrome (105). The risk of complete atrioventricular block in patients with atrioventricular discordance has been demonstrated, and SCA anomaly likely does not present an additional risk for atrioventricular block.
3.4.4. SCA with cardiomyopathyThe present study showed that 12% (7/59) of patients had coexisting cardiomyopathy, 6 had ischaemic cardiomyopathy due to CHD, and 1 had valvular heart disease. To our knowledge, there is no case of SCA with DCM to date. There were 13 patients with hyperlipidemia.
17 patients were diagnosed with SCA by computed tomography (CT). It has been reported that CTCA is the primary method for determining the diagnosis of SCA and can help delineate the course of the proximal artery. CTCA is also an interesting new model; in addition to being noninvasive, it reveals adjacent structures to understand the origin and development of coronary arteries (89).
Guidelines regarding the management of SCA are difficult to establish, and treatment is guided by symptoms and the presence or severity of atherosclerosis stenosis or the occurrence of acute coronary syndrome. The prognosis of patients ranges from a good normal life expectancy to sudden death.
4. SummaryAccording to the clinical symptoms and examination results of the patient, we believe that the patient's SCA was benign and was not the cause of his DCM. Another case of absence of the RCA with coexistence of the SCN5A gene mutation was previously reported by Katsaras D et al. (106). In this case, a patient with familial Brugada syndrome with absence of the RCA tested positive for a SCN5A gene C.664C. > T variant and presented with patent foramen ovale. In contrast, our case did not show Brugada syndrome rather than demonstrating DCM. As we discussed above, the SCN5A gene variant is associated with a spectrum of clinical diseases, and loss of sodium channel function has a critical role in the development of cardiomyopathy (48). The proband and his children carried the same three mutations. His children refused echocardiography but currently have no symptoms of DCM. The genetic background of patients presenting with DCM is complex; some studies have shown that more than 38% have a compound heterozygous or combined mutation, and 12.8% have three or more mutations (43–45). It is unclear whether the SCN5A gene mutation correlated with RCA absence and DCM in the present case. Meanwhile, the proband and his children carried an APOA5 gene mutation, which is considered a suspected pathogenic variant resulting in hypertriglyceridemia or hyperlipoproteinemia independent of the SCN5A mutation.
5. ConclusionThis is the first case of SCA combined with DCM of the SCN5A C.1858C > T (P.arg620Cys) mutation, which is the cause of DCM and Brugada syndrome. We should carefully identify and reduce life-threatening events in clinical practice to improve the survival rate. As with many other coronary artery abnormalities, coronary angiography is the gold standard method of diagnosis; importantly, CTCA plays a crucial role in diagnosis when considered as a noninvasive operation. Moreover, the underlying aetiological and pathological link between SCA and DCM remains to be explored.
Author contributionsMaterial preparation, data collection and analysis were performed by JY and XH. The first draft of the manuscript was written by JY and XH. All authors contributed to the article and approved the submitted version.
Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary materialThe Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2023.1113886/full#supplementary-material.
AbbreviationsSCA, single coronary artery; DCM, dilated cardiomyopathy; RCA, right coronary artery; CCTA, computed tomography coronary angiogram; CMR, cardiac magnetic resonance imaging; TTE, transthoracic echocardiography; LVEF, left ventricular systolic function; LVEDD, left ventricular end-diastole diameter; LVEDV, left ventricular end-diastole volume; LAD, left anterior descending; LCX, left circumflex.
References1. Cirakoglu OF, Bayraktar A, Sayin MR. An extremely rare clinical entity: congenitally corrected transposition with situs Inversus and single coronary artery presented with complete atrioventricular block in a young man. Cardiol Young. (2018) 28:759–61. doi: 10.1017/S1047951118000069
PubMed Abstract | CrossRef Full Text | Google Scholar
2. Turkmen S, Yolcu M, Sertcelik A, Ipek E, Dokumaci B, Batyraliev T. Single coronary artery incidence in 215,140 patients undergoing coronary angiography. Folia Morphol. (2014) 73:469–74. doi: 10.5603/Fm.2014.0070
CrossRef Full Text | Google Scholar
4. Zheng GM, Bai J, Tang JM, Zhu FC, Jing HX. A case of hypertrophic cardiomyopathy combined with muscular ventricular septal defect and abnormal origin of right coronary artery. Bmc Cardiovasc Disor. (2019) 19:16. doi: 10.1186/s12872-018-0997-8
CrossRef Full Text | Google Scholar
5. Pinto YM, Elliott PM, Arbustini E, Adler Y, Anastasakis A, Bohm M, et al. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: a position statement of the ESC working group on myocardial and pericardial diseases. Eur Heart J. (2016) 37:1850–8. doi: 10.1093/eurheartj/ehv727
PubMed Abstract | CrossRef Full Text | Google Scholar
6. Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, et al. Classification of the cardiomyopathies: a position statement from the European society of cardiology working group on myocardial and pericardial diseases. Eur Heart J. (2008) 29:270–6. doi: 10.1093/eurheartj/ehm342
PubMed Abstract | CrossRef Full Text | Google Scholar
7. Gerullm B, Klaassen S, Brodehl A. The genetic landscape of cardiomyopathies. Genet Causes Card Dis. (2019) 7:45–91. doi: 10.1007/978-3-030-27371-2_2
CrossRef Full Text | Google Scholar
8. Reichart D, Magnussen C, Zeller T, Blankenberg S. Dilated cardiomyopathy: from epidemiologic to genetic phenotypes A translational review of current literature. J Intern Med. (2019) 286:362–72. doi: 10.1111/joim.12944
PubMed Abstract | CrossRef Full Text | Google Scholar
10. Riuro H, Campuzano O, Berne P, Arbelo E, Iglesias A, Perez-Serra A, et al. Genetic analysis, in silico prediction, and family segregation in long QT syndrome. Eur J Hum Genet. (2015) 23:79–85. doi: 10.1038/ejhg.2014.54
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Kapplinger JD, Giudicessi JR, Ye D, Tester DJ, Callis TE, Valdivia CR, et al. Enhanced classification of brugada syndrome-associated and long-QT syndrome-associated genetic variants in the SCN5A-encoded na(v)1.5 cardiac sodium channel. Circ-Cardiovasc Gene. (2015) 8:582–95. doi: 10.1161/Circgenetics.114.000831
CrossRef Full Text | Google Scholar
12. Hoshi M, Du XX, Shinlapawittayatorn K, Liu HY, Chai S, Wan XP, et al. Brugada syndrome disease phenotype explained in apparently benign sodium channel mutations. Circ-Cardiovasc Gene. (2014) 7:123–31. doi: 10.1161/Circgenetics.113.000292
CrossRef Full Text | Google Scholar
13. Kapplinger JD, Tester DJ, Alders M, Benito B, Berthet M, Brugada J, et al. An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm. (2010) 7:33–46. doi: 10.1016/j.hrthm.2009.09.069
PubMed Abstract | CrossRef Full Text | Google Scholar
14. Jin JL, Sun D, Cao YX, Zhang HW, Guo YL, Wu NQ, et al. Intensive genetic analysis for Chinese patients with very high triglyceride levels: relations of mutations to triglyceride levels and acute pancreatitis. Ebiomedicine. (2018) 38:171–7. doi: 10.1016/j.ebiom.2018.11.001
PubMed Abstract | CrossRef Full Text | Google Scholar
15. Shah RA, Asatryan B, Dabbagh GS, Aung N, Khanji MY, Lopes LR, et al. Frequency, penetrance, and variable expressivity of dilated cardiomyopathy-associated putative pathogenic gene variants in UK biobank participants. Circulation. (2022) 146:110–24. doi: 10.1161/Circulationaha.121.058143
PubMed Abstract | CrossRef Full Text | Google Scholar
16. Argiro A, Ho C, Day SM, van der Velden J, Cerbai E, Saberi S, et al. Sex-related differences in genetic cardiomyopathies. J Am Heart Assoc. (2022) 11:e024947. doi: 10.1161/JAHA.121.024947
PubMed Abstract | CrossRef Full Text | Google Scholar
17. Andersson C, Schou M, Schwartz B, Vasan RS, Christiansen MN, D’Souza M, et al. Incidence rates of dilated cardiomyopathy in adult first-degree relatives versus matched controls. Int J Cardiol Heart Vasc. (2022) 41:101065. doi: 10.1016/j.ijcha.2022.101065
PubMed Abstract | CrossRef Full Text | Google Scholar
18. Miura K, Nakagawa H, Morikawa Y, Sasayama S, Matsumori A, Hasegawa K, et al. Epidemiology of idiopathic cardiomyopathy in Japan: results from a nationwide survey. Heart. (2002) 87:126–30. doi: 10.1136/heart.87.2.126
PubMed Abstract | CrossRef Full Text | Google Scholar
22. Keller H, Finsterer J, Steger C, Wexberg P, Gatterer E, Khazen C, et al. Novel c.367_369del LMNA mutation manifesting as severe arrhythmias, dilated cardiomyopathy, and myopathy. Heart Lung. (2021) 41:382–6. doi: 10.1016/j.hrtlng.2011.07.007
CrossRef Full Text | Google Scholar
23. Gaertner A, Klauke B, Felski E, Kassner A, Brodehl A, Gerdes D, et al. Cardiomyopathy-associated mutations in the RS domain affect nuclear localization of RBM20. Hum Mutat. (2020) 41:1931–43. doi: 10.1002/humu.24096
PubMed Abstract | CrossRef Full Text | Google Scholar
24. Brodehl A, Dieding M, Biere N, Unger A, Klauke B, Walhorn V, et al. Functional characterization of the novel DES mutation p.L136P associated with dilated cardiomyopathy reveals a dominant filament assembly defect. J Mol Cell Cardiol. (2016) 91:207–14. doi: 10.1016/j.yjmcc.2015.12.015
PubMed Abstract | CrossRef Full Text | Google Scholar
25. Lukas Laws J, Lancaster MC, Ben Shoemaker M, Stevenson WG, Hung RR, Wells Q, et al. Arrhythmias as presentation of genetic cardiomyopathy. Circ Res. (2022) 130:1698–722. doi: 10.1161/CIRCRESAHA.122.319835
PubMed Abstract | CrossRef Full Text | Google Scholar
26. Herman DS, Lam L, Taylor MR, Wang L, Teekakirikul P, Christodoulou D, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. (2012) 366:619–28. doi: 10.1056/NEJMoa1110186
PubMed Abstract | CrossRef Full Text | Google Scholar
27. Taylor MR, Slavov D, Ku L, Di Lenarda A, Sinagra G, Carniel E, et al. Prevalence of desmin mutations in dilated cardiomyopathy. Circulation. (2007) 115:1244–51. doi: 10.1161/CIRCULATIONAHA.106.646778
PubMed Abstract | CrossRef Full Text | Google Scholar
28. Schmitt JP, Kamisago M, Asahi M, Li GH, Ahmad F, Mende U, et al. Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban. Science. (2003) 299:1410–3. doi: 10.1126/science.1081578
PubMed Abstract | CrossRef Full Text | Google Scholar
29. Gerull B, Gramlich M, Atherton J, McNabb M, Trombitas K, Sasse-Klaassen S, et al. Mutations of TTN, encoding the giant muscle filament titin, cause familial dilated cardiomyopathy. Nat Genet. (2002) 30:201–4. doi: 10.1038/ng815
PubMed Abstract | CrossRef Full Text | Google Scholar
30. Shen C, Xu L, Sun X, Sun A, Ge J. Genetic variants in Chinese patients with sporadic dilated cardiomyopathy: a cross-sectional study. Ann Transl Med. (2022) 10:129. doi: 10.21037/atm-21-6774
PubMed Abstract | CrossRef Full Text | Google Scholar
31. Sarica AS, Bor S, Orman MN, Barajas-Martinez H, Juang JJ, Antzelevitch C, et al. Frequency of irritable bowel syndrome in patients with brugada syndrome and drug-induced type 1 brugada pattern. Am J Cardiol. (2021) 151:51–6. doi: 10.1016/j.amjcard.2021.04.010
PubMed Abstract | CrossRef Full Text | Google Scholar
32. Pan X, Li Z, Jin X, Zhao Y, Huang G, Huang X, et al. Comparative structural analysis of human Nav1.1 and Nav1.5 reveals mutational hotspots for sodium channelopathies. Proc Natl Acad Sci USA. (2021) 118:e2100066118. doi: 10.1073/pnas.2100066118
CrossRef Full Text | Google Scholar
33. Strege PR, Mazzone A, Bernard CE, Neshatian L, Gibbons SJ, Saito YA, et al. Irritable bowel syndrome patients have SCN5A channelopathies that lead to decreased Na1.5 current and mechanosensitivity. Am J Physiol Gastrointest Liver Physiol. (2018) 314:G494–G503. doi: 10.1152/ajpgi.00016.2017
PubMed Abstract | CrossRef Full Text | Google Scholar
34. Verstraelen TE, Ter Bekke RM, Volders PG, Masclee AA, Kruimel JW. The role of the SCN5A-encoded channelopathy in irritable bowel syndrome and other gastrointestinal disorders. Neurogastroenterol Motil. (2015) 27:906–13. doi: 10.1111/nmo.12569
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Neshatian L, Strege PR, Rhee PL, Kraichely RE, Mazzone A, Bernard CE, et al. Ranolazine inhibits voltage-gated mechanosensitive sodium channels in human colon circular smooth muscle cells. Am J Physiol Gastrointest Liver Physiol. (2015) 309:G506–12. doi: 10.1152/ajpgi.00051.2015
PubMed Abstract | CrossRef Full Text | Google Scholar
36. Strege PR, Ou Y, Sha L, Rich A, Gibbons SJ, Szurszewski JH, et al. Sodium current in human intestinal interstitial cells of Cajal. Am J Physiol Gastrointest Liver Physiol. (2003) 285:G1111–21. doi: 10.1152/ajpgi.00152.2003
PubMed Abstract | CrossRef Full Text | Google Scholar
37. Poller W, Escher F, Haas J, Heidecker B, Schultheiss HP, Attanasio P, et al. Missense variant E1295K of sodium channel SCN5A associated with recurrent ventricular fibrillation and myocardial inflammation. JACC Case Rep. (2022) 4:280–6. doi: 10.1016/j.jaccas.2022.01.016
PubMed Abstract | CrossRef Full Text | Google Scholar
38. Jones A, Kainz D, Khan F, Lee C, Carrithers MD. Human macrophage SCN5A activates an innate immune signaling pathway for antiviral host defense. J Biol Chem. (2014) 289:35326–40. doi: 10.1074/jbc.M114.611962
PubMed Abstract | CrossRef Full Text | Google Scholar
39. Rahgozar K, Wright E, Carrithers LM, Carrithers MD. Mediation of protection and recovery from experimental autoimmune encephalomyelitis by macrophages expressing the human voltage-gated sodium channel NaV1.5. J Neuropathol Exp Neurol. (2013) 72:489–504. doi: 10.1097/NEN.0b013e318293eb08
PubMed Abstract | CrossRef Full Text | Google Scholar
41. Wilde AAM, Amin AS. Clinical spectrum of SCN5A mutations: long QT syndrome, brugada syndrome, and cardiomyopathy. JACC Clin Electrophysiol. (2018) 4:569–79. doi: 10.1016/j.jacep.2018.03.006
PubMed Abstract | CrossRef Full Text | Google Scholar
42. Ge J, Sun A, Paajanen V, Wang S, Su C, Yang Z, et al. Molecular and clinical characterization of a novel SCN5A mutation associated with atrioventricular block and dilated cardiomyopathy. Circ Arrhythm Electrophysiol. (2008) 1:83–92. doi: 10.1161/CIRCEP.107.750752
PubMed Abstract | CrossRef Full Text | Google Scholar
43. Huang W, Xu R, Gao N, Wu X, Wen C. Corrigendum: case report: family curse: an SCN5A mutation, c.611C>A, p.A204E associated with a family history of dilated cardiomyopathy and arrhythmia. Front Cardiovasc Med. (2022) 9:944834. doi: 10.3389/fcvm.2022.944834
PubMed Abstract | CrossRef Full Text | Google Scholar
44. Haas J, Frese KS, Peil B, Kloos W, Keller A, Nietsch R, et al. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur Heart J. (2015) 36:1123–35a. doi: 10.1093/eurheartj/ehu301
PubMed Abstract | CrossRef Full Text | Google Scholar
45. Akinrinade O, Ollila L, Vattulainen S, Tallila J, Gentile M, Salmenpera P, et al. Genetics and genotype-phenotype correlations in Finnish patients with dilated cardiomyopathy. Eur Heart J. (2015) 36:2327–37. doi: 10.1093/eurheartj/ehv253
PubMed Abstract | CrossRef Full Text | Google Scholar
46. Mann SA, Castro ML, Ohanian M, Guo G, Zodgekar P, Sheu A, et al. R222q SCN5A mutation is associated with reversible ventricular ectopy and dilated cardiomyopathy. J Am Coll Cardiol. (2012) 160:566–73. doi: 10.
留言 (0)