

記住我
The third most common cause of cancer-related death worldwide, gastric cancer (GC) is one of the most prevalent malignant tumors. The prevalence of GC varies greatly among various geographical areas (Machlowska et al., 2020; Petryszyn et al., 2020). Its growth and evolution are multi-year, multi-stage processes that continue to be a problem for global health today (Gao et al., 2018).
The most significant risk factor for GC is Helicobacter pylori (H. pylori), which is one of the most prevalent infectious organisms in humans globally (Noto and Peek, 2012). H. pylori is categorized by the World Health Organization (WHO) as a class 1 carcinogen (Crowe, 2019). Numerous virulence factors produced by H. pylori have the potential to disrupt intracellular signaling pathways in the host and lower the threshold for tumor transformation. In addition, gastric cancer, the H. pylori infection can also cause other stomach diseases, including gastric ulcer, duodenal ulcer, stomach atrophy and other diseases. The H. pylori infection is strongly associated with major gastritis and multifocal atrophy, and testing for H. pylori is another important component of screening for these diseases.
The main pathogenic agents in the H. pylori infection are CagA (cytotoxin-related gene A), its pathogenicity island (Cag PAI), and VacA (vacuolar cytotoxin A) (Wang F et al., 2014). The CagA protein and the Cag IV secretion system (T4SS) are encoded by 27–31 genes in the 40 kb Cag PAI DNA insertion element (Muller, 2012; Backert et al., 2015). The three proteins CagL, CagI, and CagH are parts of the T4SS subcomponents and are all necessary for Cag T4SS, which may be crucial in the development of the infectious contact between H. pylori and stomach epithelial cells. Through bacteria and epithelial cells, CagA T4SS transports CagA from connected H. pylori into host cells, through the passage of bacterial and epithelial membranes, CagA T4SS carries CagA from the associated into the host cells (Odenbreit et al., 2000; Fischer et al., 2001; Shaffer et al., 2011). It binds to the inside of cell membranes, causing the downstream signaling pathways to be activated by tyrosine phosphorylation at the N-terminal glutamate-proline-isoleucine-tyrosine-alanine (EPIYA) site (Wroblewski and Peek, 2016). Vacuolar cytotoxin A (VacA), an 88 kDa protein made up of the p33 and p55 protein subunits, can be secreted using the IV-type autotransporter and secretion system. This protein alters a number of things, including the permeability of the mitochondrial membrane, the vacuolation of host gastric epithelial cells, autophagy, apoptosis, and disruption of epithelial tight junctions. Additionally, it prevents lamina propria T lymphocyte activation and proliferation (Cover and Blanke, 2005; Palframan et al., 2012; Raju et al., 2012).
The release of virulence factors following the H. pylori infection of gastric epithelial cells can activate downstream signaling pathways and associated processes, including the nuclear factor κB (NF-κB) pathway and the cytokine-stimulated transduction (JAK-STAT) signaling system. Furthermore, these virulence factors have the ability to induce apoptosis and methylation of the relevant proteins, which can control the expression of a number of host proteins and influence the appearance and growth of GC.
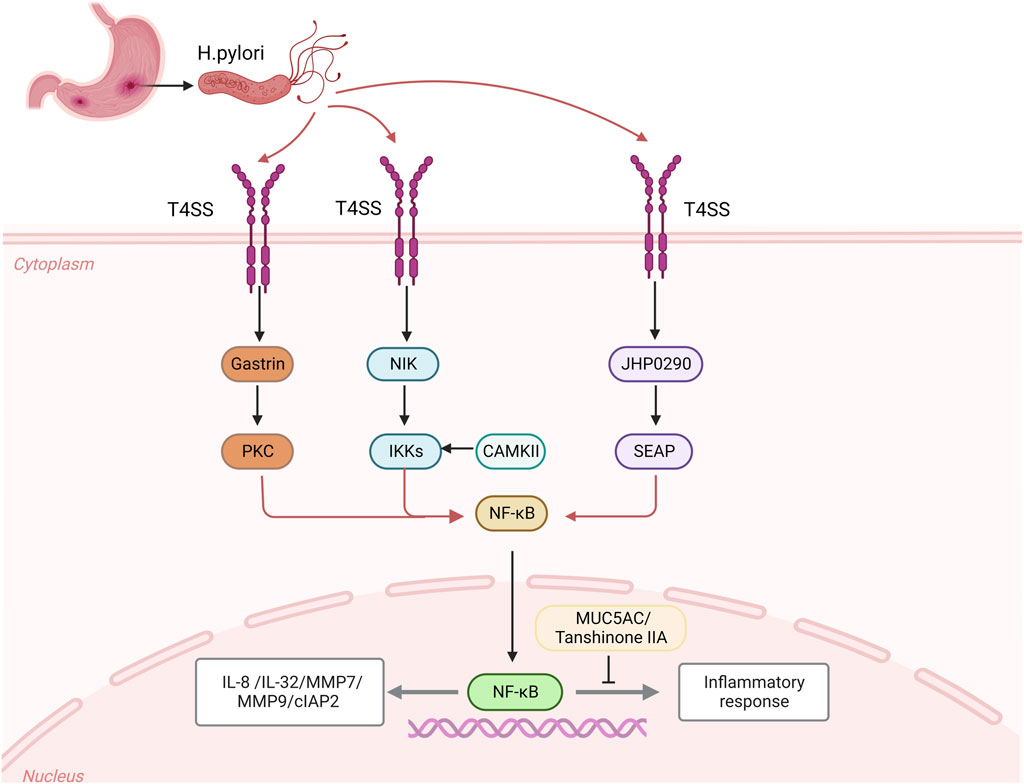
Helicobacter pylori infected host cells activate NF-κB signaling pathwayGastric epithelial cells were colonized by H. pylori, which then activated the natural and NF-κB pathway (Maubach et al., 2021; Maubach et al., 2022). Inflammation is brought on by H. pylori CagA stimulating the NF-κB pathway and binding TAK1 to TRAFs, which in turn activates the IκB kinase (IKK) complex (Brandt et al., 2005; Zhang et al., 2019). Protein modification and intracellular location control the formation of homologous dimers and heterodimers that both activate and inhibit transcription (Neumann and Naumann, 2007), the NF-κB family of transcription factors regulates immunological response, inflammatory response (Hayden and Ghosh, 2011), cell proliferation, differentiation, and genomic stability (Liu et al., 2017; Peng et al., 2020).
Following the H. pylori infection, GC cells secreted more IL-8 and IL-32. For the IL-8 gene to be transcribed, the essential transcription factor NF-κB must be activated by binding to either AP-1 or NF-IL6 (Yasumoto et al., 1992), in GC cells, AP-1 may take the place of NF-IL6 and work in conjunction with NF-κB to cause IL-8 gene transcription binding to form a complex (Aihara et al., 1997). Additionally, NF-κB increases the expression of the inflammatory cytokine IL-32, which in turn increases the expression of NF-κB. This occurs mostly through the CAG pathogenicity island (cagPAI)-positive (Sakitani et al., 2012). A crucial metalloproteinase is MMP-7, in a CAG pathogenicity island (cagPAI) -dependent way, causes a persistent inflammatory response and increases gastrin expression in gastric epithelial cells. Gastrin increases MMP-7 via triggering the NF-κB signaling pathway via the protein kinase C-dependent pathway linked to IκB kinase. Additionally, it stimulates the expression of the HB-EGF gene for epidermal growth factor and ectodomain shedding. (Bebb et al., 2003; Ogasa et al., 2003; Dickson et al., 2006; Yin et al., 2010). Additionally, a the H. pylori infection of the stomach causes the release of MMP-9, a matrix metalloproteinase (Hojo et al., 2000; Göõz et al., 2001), the intracellular kinases NIK and IKKs stimulate the NF-κB signaling pathway in gastric epithelial cells infected with cagPAI-positive H. pylori, controlling the production of MMP-9 (Maeda et al., 2000; Mori et al., 2003). The virulence factor urease and the effects of H. pylori on the expression and transcription levels of MUC in MUC genes (MUC5AC, MUC2, and MUC6) in gastric mucosa of Kato-III may upregulate the expression of chemokines and proinflammatory factors while downregulating the transcription of the MUC5AC gene in GC cells. On the other hand, the MUC5AC promoter has a κB cis-element, which reduces the activity of the promoter and lowers expression (Perrais et al., 2014). MUC1 is essential for controlling how negatively NLRP3 inflammasome activation affects immune cells. MUC1 expression rises following the H. pylori infection, which prevents NLRP3 from being activated by blocking the TLR/NF-κB-dependent signaling pathway. This, in turn, prevents the inflammatory response brought on by chronic the H. pylori infection (Ng et al., 2016; Zhang et al., 2022a).
One of the primary proteins regulating apoptosis is inhibitor of apoptosis protein (IAP), and cIAP2 (inhibitor of Apoptosis protein 2) is crucial for the development of cancer (Chen et al., 2020). The advancement of AG/IM and the H. pylori infection are not the only conditions that are linked to the overexpression of cIAP2 in GC, the primary mechanism is that H. pylori upregulates the expression of cIAP2 by activating the NF-κB signaling pathway in a cagPAI dependent manner (Yoon et al., 2017). A homolog of HP0305 called JHP0290 can attach to different cell types and alter macrophage reactions (Pathak et al., 2013). Different H. pylori strains can express and release the JHP0290 homolog. NF-κB activation was seen in gastric epithelial cells that had been stimulated by JHP0290, which considerably increased the amount of alkaline phosphatase (SEAP) activity in GC cells in a dose-dependent manner and activated the NF-κB signaling pathway, which controls a number of cellular processes in cancer (Tavares and Pathak, 2015). Additionally, H. pylori stimulates the LIGHT pathway, a distinct group of receptors in the tumor necrosis factor superfamily, in addition to the canonical NF-κB signaling pathway, which necessitates functional T4SS (TNFSF). The main mechanism is that the CAG pathogenicity island (cagPAI), after infecting gastric epithelial cells with H. pylori, stimulates ligand binding to the LTβR produced by epithelial cells and draws in immune cells to increase chemokines and suppress the usual NF-κB signaling pathway. There is a close connection between the common and alternate pathways during the H. pylori infection (Mejias-Luque et al., 2017). The expression of CAMKII (Ca21/calmodulin dependent kinase II) is regulated by calmodulin, the IKK complex is activated by CAMKII and calmodulin, which triggers the NF-κB signaling pathway (Maubach et al., 2013). Additionally, after the H. pylori infection, several medical substances can help control how proteins are expressed. Tanshinone IIA can effectively inhibit the activation of the NF-κB signaling pathway, destroying the production of downstream inflammatory substances and effectively reducing the inflammatory response induced by H. pylori. After the H. pylori infection, the expression of nuclear NF-κB (p65) protein increases significantly (Chen et al., 2016) (Figure 1).
FIGURE 1. After the Helicobacter pylori infection, CagA is transferred from the attached H. pylori through the bacteria and epithelial cells to the host cell through the CagA-T4SS secretion system, which activates the expression of intracellular kinases NIK and gastrin as well as the homologues of JHP0290, and activates the activities of downstream intracellular kinases IKKs, PKC and SEAP, activate the NF-κB signaling pathway to regulate the expression of inflammatory cytokines IL-8 and IL-32, MMP7 and MMP9, and apoptosis inhibitor protein 2 (cIAP2). CAMKII and calmodulin can activate IKK complex and induce NF-κB signal. After the H. pylori infection, MUC5AC could bind to κB cil-element promoter, which caused decreases of MUC5AC promoter’s activity and expression of MUC5AC. The release of Tanshinone IIA after the H. pylori infection can effectively inhibit the expression of NF-κB nuclear protein and inhibit the inflammatory signal.
Helicobacter pylori infected host cells activate ERK/MAPK signaling pathwayThe MAPK signaling pathway can be activated by H. pylori-induced gastric epithelial cell growth and gene expression (Sebkova et al., 2004; Zhu et al., 2005; Ding et al., 2008). The mitogen-activated protein kinase (MAPK) family, which participates in signaling cascades and transmits extracellular signals to intracellular destinations, includes extracellular signal-regulated kinase 1/2 (ERK). In eukaryotic cells: ERK, JNK/stress-activated protein kinase, P38 MAPK, and ERK5 signal transduction pathways have all been found (Guo et al., 2020b). The extracellular signal-regulated protein kinase (ERK) cascade, which is typically controlled by the activation of cell-surface receptor tyrosine kinases (RTKS), is the most prevalent of these (Katz et al., 2007). A fundamental signal transduction system known as the MAPK signaling pathway controls cell growth, differentiation, and stress response (Plotnikov et al., 2011; Sabio and Davis, 2014; Guo et al., 2020b).
AUF1 is upregulated when H. pylori is present as a result of CagA-induced ERK pathway activation. GKN1 mRNA, a gastric tumor suppressor, can have its stability controlled by the ARE-binding protein AUF1. In light of this, it is possible that the CagA/p-ERK/AUF1 axis is crucial in the downregulation of the downstream AUF1 effector GKN1mRNA. Making it a GC oncogene (Guo et al., 2020a). Through the NF-κB signaling pathway, H. pylori can cause the expression of metalloproteinases, and through the ERK signaling pathway, it can control the expression of metalloproteinases. The H. pylori CAG pathogenicity island (cagPAI), which is implicated in GC metastasis, induces the ERK1/2 signaling pathway, which is responsible for the upregulation of MMP-1 production in gastric epithelial cells (Krueger et al., 2006; Jiang et al., 2014). The ERK1/2 signaling pathway activated by H. pylori also controls MMP-10. The CAGA-positive H. pylori strain damages the stomach epithelium by prompting gastric epithelial cells to produce and secrete active MMP-10 in GC cells, which in turn activates the tyrosine kinase receptor EGFR (Costa et al., 2016). Additionally, the MMP-10 expression caused by H. pylori was reduced by the red-orange pigment β-carotene, which in vivo can be converted to retinaldehyde, mostly by activating PPAR-γ and triggering its downstream target catalase. As a result, H. pylori-infected gastric epithelial cells have lower levels of ROS and the ERK signaling pathway, as well as lower levels of MMP-10 expression and H. pylori-associated GC incidence (Costa et al., 2016; Bae et al., 2021).
The alteration in cell behavior brought on by H. pylori is mediated by AQP3. The expression of AQP3 in gastric cells is primarily controlled by the ERK signaling pathway, and reduction of AQP3 can inhibit the proliferation and migration of cancer cells generated by H. pylori. An essential membrane protein called Cxs controls the creation of intercellular channels, the interchange of signaling chemicals, and intercellular communication. Additionally, it plays a crucial role in intercellular communication (Vinken et al., 2006; Gemel et al., 2014). The virulence factor of H. pylori, the fact that VacA has no effect on the membrane protein Cx43’s mRNA level raises the possibility that VacA might promote Cx43 accumulation by preventing Cx43 degradation. The amount of GSH that is turned over in GC cells may be controlled by VacA released by H. pylori (Kimura et al., 2001); the GSH levels influence the ROS-dependent ERK signaling pathway’s activity, which controls the generation of Cx43 and apoptosis (Yahiro et al., 2015). Furthermore, H. pylori JHP0290 protein can not only activate the NF-κB pathway to participate in the inflammatory response, but also activate ERK MAPK in a dose-dependent manner to regulate the proliferation of gastric epithelial cells (Tavares and Pathak, 2015) (Figure 2).
FIGURE 2. After the H. pylori infection, virulence factors are secreted into cells through the CagA-T4SS secretion system, activation of ROS and Ras/Raf/MEK proteins, activation of downstream ERK/MAPK signaling pathway, upregulate of AUF1 and control of GKN1 expression, ERK also control the expression of metal proteinases MMP1 and MMP10. β-carotene inhibits H. pylori induced the expression of MMP10, and the H. pylori virulence factor can upregulate Cx43 expression, and then activate the ROS-dependent ERK signaling pathway to reverse control Cx43 production and apoptosis. AQP3 is involved in the changes of cell behavior induced by H. pylori through the regulation of ERK pathway. JHP0290 released by H. pylori protein can activate the ERK pathway and regulate the proliferation of gastric epithelial cells.
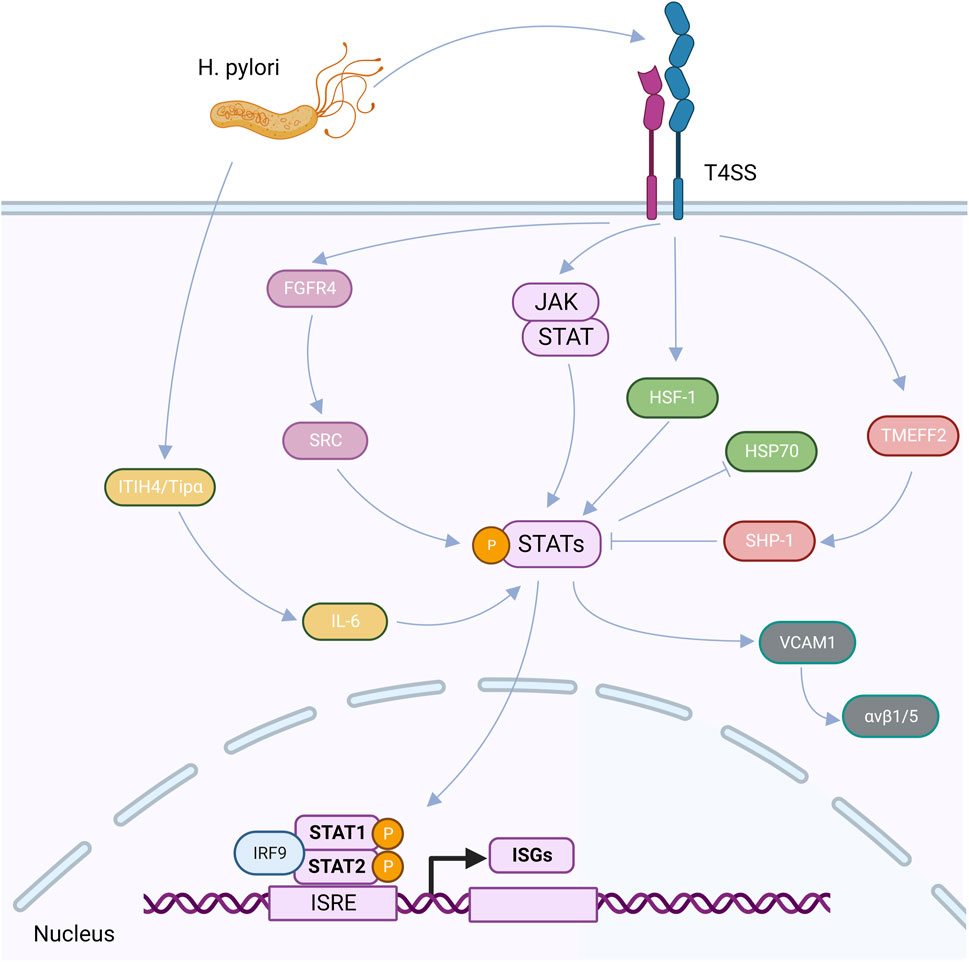
Helicobacter pylori infected host cells activate JAK/STAT signaling pathwayThrough the JAK/STAT signaling system, the H. pylori infection of gastric epithelial cells can control cell growth and the production of associated proteins. An essential signal transduction system is the JAK/STAT (Janus kinase/Signal Converter and Activator of transcription) cascade (Khanna et al., 2015). To control the expression of the associated genes, JAK phosphorylates STAT, which dimerizes and travels to the nucleus through the nuclear envelope (Xin et al., 2020). A family of non-transmembrane tyrosine kinases is known as the JAK family. The JAK clan is Most members of the JAK family are JAK1, JAK2, JAK3, and Tyk2 (Cai et al., 2015). One of the most important activating transcription factors in the immune response is the STAT family, which is a downstream target of JAKs in the cytoplasm. There are seven people who make up the group: STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, and STAT6 (Boengler et al., 2008; Yu et al., 2009), cell proliferation, stem cell self-renewal, and immunological responses are only a few examples of the physiological and cellular processes that JAK/STAT signaling is involved in that are connected to the beginning and development of disease (Aaronson and Horvath, 2002; Rawlings et al., 2004).
Human inter-trypsin inhibitor heavy chain 4 (ITIH4) is an acute phase response protein that is positively regulated by interleukin-6 (IL-6) (Piñeiro et al., 1999; Liu et al., 2016), and TIP-α (tumor necrosis factor-α-inducible protein), a newly identified membrane protein secreted by H. pylori, is a potent inducer of epithelial-mesenchymal transition (EMT). After the H. pylori infection, the secretion of ITIH4 and TIP-αmay encourage the production of IL-6 in GC cells, IL-6 can then trigger the expression of p-STAT3, which activates the STAT3 signaling pathway. The IL-6/STAT3 pathway may be activated by Tip-α and ITIH4 to speed up GC (Jove, 2000; Liu et al., 2016; Chen et al., 2017; Suganuma et al., 2021). TMEFF2 is a signaling transmembrane protein that interacts with two folliclestatin proteins and epidermal growth factor (Costa et al., 2010; Lin et al., 2011). TMEFF2 in the GC may not be regulated normally as a result of the H. pylori infection. The primary mechanism is that, in the early stages of the H. pylori infection, the overexpression of TMEFF2 in healthy gastric mucosa causes the production of SHP-1, a protein tyrosine phosphatase, which inhibits STAT3 activation. A long-lasting the H. pylori infection can activate the STAT3 signaling pathway, control STAT3 phosphorylation, and bind directly to the TMEFF2 promoter to suppress TMEFF2 production in the opposite direction (Sun et al., 2015).
High FGFR4 expression and STAT3 activation levels can result from the H. pylori infection. SRC serves as a bridge between STAT3 and FGFR4, indicating that STAT3 is involved in the stimulation of FGFR4 signaling and demonstrating a positive feedback loop between STAT3 and FGFR4 (Zhang et al., 2022b). Heat shock factor 1 (HSF-1) and phosphorylated STAT-3 interact at the protein level after the H. pylori infection of GC cells to produce transcriptionally inactivated HSF-1/STATs complex, which inhibits HSP70 expression, loses its cytoprotective function, and becomes less vulnerable to apoptosis induction (Pierzchalski et al., 2006). A significant stromal component of many types of malignancies are cancer-associated fibroblasts (CAFs) (Su et al., 2018), which express smooth muscle actin (α-SMA), fibroblast activating protein, and fibroblast specific protein 1 (FSP-1) (Kalluri, 2016). By triggering the JAK/STAT1 signaling system, a gastric the H. pylori infection can increase the expression of vascular adhesion molecule 1 (VCAM1) in fibroblasts. By connecting with integrin αVβ1/5, VCAM1 can facilitate GC cells’ infiltration into tumors (Shen et al., 2020) (Figure 3).
FIGURE 3. After the H. pylori infection, virulence factors can be secreted into cells through the CagA-T4SS secretion system, upregulated ITIH4 and TiP-α, and induced the high expression of IL-6, thus activating the expression of p-STAT3, that is, the STAT3 signaling pathway is activated. After the H. pylori infection, TMEFF2 induced upregulation of SHP-1 and inhibited STAT3 phosphorylation. After the H. pylori infection, FGFR4 was highly expressed, which upregulated SRC and activated STAT signaling pathway. After the H. pylori infection, HF-1 interacts with phosphorylated STAT-3, resulting in the suspension of HSP70 expression. After the H. pylori infection, VCAM1 can be upregulated by activating the JAK/STAT1 signaling pathway, and then interact with integrin αvβ1/5 to promote tumor invasion. After the activation of STAT signaling pathway, IRF9 can interact with homologous dimers formed by STAT1 and STAT2 on functional IFN stimulatory regulatory elements (ISRE) in the nucleus to regulate the transcription and expression of ISGs.
Helicobacter pylori infection induces host cell apoptosisAn active physiological process of cell death is known as apoptosis. By releasing virulence factors, the H. pylori infection can activate and regulate the associated proteins, leading to the death of gastric epithelial cells (Steller, 1995; Ashktorab et al., 2008). Caspases are the aspartate cysteine protease family’s proteasomes, and their activation is typically required for apoptosis to occur (Cohen, 1997), caspases -3, -6, -8, -9, and -10, for example, like caspase-3 or caspase-6, caspase-8 is a promoter protease (Sun et al., 2006). The Bcl-2 family is also a sizable collection of proteins linked to apoptosis. In response to varied stimuli, the balance of pro- and anti-apoptotic molecules determines whether the cells survive or perish. Among them, Bcl-2, Bax, and Bcl-XL control the essential proteins of cell death, mostly through influencing mitochondrial function and encouraging the release of cytochrome C (Guo et al., 2019; Li et al., 2020; Wu et al., 2020; Yi et al., 2020; Liu et al., 2021). H. pylori can regulate the downstream apoptotic factor caspase and the expression of related proteins of apoptosis-related genes p53, Bax, and Fas pathways by activating mitochondria to release cytochrome, triggering the expression of various cell substrates and leading to cell apoptosis by interacting with death receptors in the serous membrane.
Reactive oxygen species (ROS) can cause Ape-1, a multifunctional protein that regulates apoptosis, to be produced (Ramana et al., 1998). In the exogenous pathway leading to the gastric epithelial cells’ programmed death after the H. pylori infection, APE-1 acetylation is a critical component. Acetylation mutants overexpressing APE-1 prevented caspase-9 activation after the H. pylori infection, resulting in decreased expression. The ability of the caspase-8-mediated apoptotic pathway was diminished concurrently (Chattopadhyay et al., 2010). The src-and C-Abl (non-receptor tyrosine kinases)-mediated phosphorylation of CagA is necessary for an H. pylori-mediated cell infection (Poppe et al., 2007; Mueller et al., 2012), patients with H. pylori-associated gastritis had significantly higher levels of C-Abl in the gastric epithelium and gland. PKC has the ability to directly phosphorylate pAblT735 in gastric epithelial cells. The 14-3-3 protein binds to C-Abl, pushing it to localize in the cytoplasm and inhibiting lowering the production of caspase-8 and caspase-9, blocking the intrinsic apoptotic pathway, and the caspase promoter (Raina et al., 2005; Maiani et al., 2011; Posselt et al., 2019). Patients who have been diagnosed with H. pylori have higher levels of IL-18 and IFN-γ, IFN-γ promotes the cellular synthesis of IL-18, by boosting the production of caspase-3, the intracellular cysteine protease ICE (caspase-1) contributes to the processing of IL-18’s active form and to the induction of apoptosis in gastric epithelial cells (Shimada et al., 2008; Koch and Muller, 2015). TRAF1, a member of the TRAF family, interacts with various tumor necrosis factor receptors (TNFR) directly or indirectly and inhibits apoptosis in cells by triggering inflammatory pathways. TRAF1 upregulation can be brought on in gastric epithelial cells by H. pylori. The virulence factor CagA can prevent the lysis of TRAF1 and the activation of caspase-8 to TRAF1 following the H. pylori infection. Consequently, H. pylori can persist in the H. pylori can therefore persist in the stomach mucosa for a long time without leading to apoptosis (Wan et al., 2016). By inhibiting FLIP, promoting DISC (death signal inducing complex) assembly by FLIP, activating caspase-8, transmitting the apoptotic signal to the mitochondria, and inducing the release of cytochrome C from the mitochondria into the cytoplasm, H. pylori induced TRAIL (tumor necrosis factor-associated apoptosis-inducing ligand) apoptosis signal. Apoptosis resistance can break down when the mitochondrial downstream caspase cascade caspase-9 is triggered (Lin et al., 2014).
After the H. pylori infection, corticosteroids, which activate the actin-related protein complex ARP2/3, can help the acid activate the apoptotic function in gastric epithelial cells (VacA), which in turn causes the pro-apoptotic protein Bax to be induced and the anti-apoptotic protein Bcl-2 to be inhibited. This finally causes target cells to undergo apoptosis (Chang et al., 2016). Heat shock proteins (HSPs) serve as molecular chaperones that help damaged proteins refold and fold freshly generated cellular proteins. The GC epithelial cells of H. pylori are directly affected by the production of CagA and vacA, increasing their fragility and suppressing the expression of HSP70. On the other hand, the balance between Bax and Bcl-2 expression was altered by the downregulation of HSP70 and the absence of its protective effect on the cell defense system.
Additionally, at the same time, Bcl-2, an anti-apoptotic protein, showed decreased expression (Pierzchalski et al., 2006; Targosz et al., 2012). Through pathways that depend on VDAC and the Bcl-2 family and cytochrome C, the H. pylori virulence factor VacA induces caspase-3, causing the activation of caspase-3 and the execution of apoptosis (Lan et al., 2010).
Helicobacter pylori infection induces DNA methylation in host cellsThe most extensively researched epigenetic alteration, DNA methylation, has two primary modification mechanisms. The first step is to modify the cytosine residue of the CpG dinucleotide by adding a methyl group to its fifth carbon (Feinberg and Tycko, 2004). The second is the gc-rich region of the genome’s CpG dinucleotide cluster CpG island (CGI), where abnormal methylation results in transcriptional silence and alters the expression of downstream genes (Qu et al., 2013; Usui et al., 2021). Two DNA methylation transferases (DNMTs) catalyze cytosine methylation (Jones and Baylin, 2002), including DNMT3 family methylases, which participate in de novo methylation, and DNMT1, which maintains methylation by methylating newly synthesized DNA strands (Costello and Plass, 2001; Hashimoto et al., 2010). One of the primary causes of carcinogenesis is the methylation-induced silence of tumor suppressor genes (Vogelstein et al., 2013).
Connexins Cx32 and Cx43 connect the space between gastric epithelial cells, and the H. pylori infection may result in high levels of methylation of their promoters, which lowers their expression. This inhibits the intercellular communication (GJIC) function of the gastric space junction, which causes GC. The expression of Cx32 and Cx43 decreased during the H. pylori infection’s chronic atrophic gastritis stage (Wang Y et al., 2014). In GC, FOXD3 is a tumor suppressor that has been epigenetically silenced. Once a FOXD3 promoter methylation is induced at the transcription start point following the H. pylori infection. The tumor suppressor role of FOXD3 in GC was later identified, and its direct transcriptional targets CYFIP2 and RARB may act as a conduit for this role. In human GC, FOXD3 prevents the tumor cascade from being downregulated (Cheng et al., 2013). Mucous metaplasia, or TFF2, primarily arises from the stomach’s bottom. The peptide for muscle crack is known as express solution IM (Soutto et al., 2015). The main mechanism of the H. pylori infection in chronic TFF2 promoter methylation-induced GC cells directly, primarily in the transcription start site of overlapping CpG dinucleotide, is infected cells after startup TFF2 methylation. Consequently, TFF2 expression declines over time (Peterson et al., 2010). A key regulator of the production of autophagosomes is the microtubule-associated protein 1 light chain 3 (MAP1LC3/LC3) (Ravikumar et al., 2010). MAP1LC3Av1 methylation silencing, which is mostly controlled by DNA methylation in its promoter region, can result from a long-term infection of gastric epithelial cells with H. pylori. This can result in an autophagy pathway of cell carcinogenesis. These findings imply that preventing GC brought on by H. pylori-associated epigenetic autophagy damage can be accomplished by utilizing demethylating drugs (Muhammad et al., 2017).
DiscussionThe modulation of host proteins in gastric epithelial cells infected with H. pylori has been the subject of several investigations. The primary processes of signaling pathways are reviewed in this article, as well as how signaling pathways control the expression of host proteins in GC. For instance, secreted virulence factors regulate the expression of downstream target proteins, activate NF-κB, ERK/MAPK, JAK/STAT, and other signaling pathways or cytokine receptors, enhance or inhibit the inflammatory response after infection, and promote the proliferation and metastasis of GC. Following the H. pylori infection, the expression of associated proteins and inflammatory factors is either up- or downregulated, activating the intracellular apoptosis program, starting or stopping the expression of apoptotic proteins, and controlling the associated GC process. Additionally, the H. pylori infection can result in DNA methylation, which silences the associated tumor suppressor genes and promotes the growth and spread of cancer.
Despite a thorough examination of the literature, the relevant signaling pathways—such as the PI3K-AKT pathway, Wnt/β-Catenin signaling network, and TGF-β signaling pathway—presented are not all-inclusive. It is vital to pay attention to the linked host proteins because they influence the onset and prognosis of gastric illnesses brought on by the H. pylori infections.
Author contributionsYX and X-LZ conceived the article and completed the first draft. Q-XL, H-NG, and Y-SL revised and polished the article. S-HS and X-HM directed the structure and content of the thesis and provided funding. All authors read and approved the manuscript.
FundingThis study was funded by the National Natural Science Foundation of China (Grant No. 81772157).
AcknowledgmentsThe figures in the manuscript were created with BioRender.com.
Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
ReferencesAihara, M., Tsuchimoto, D., Takizawa, H., Azuma, A., Wakebe, H., Ohmoto, Y., et al. (1997). Mechanisms involved in Helicobacter pylori-induced interleukin-8 production by a gastric cancer cell line, MKN45. Infect. Immun. 65 (8), 3218–3224. doi:10.1128/IAI.65.8.3218-3224.1997
PubMed Abstract | CrossRef Full Text | Google Scholar
Ashktorab, H., Dashwood, R. H., Dashwood, M. M., Zaidi, S. I., Hewitt, S. M., Green, W. R., et al. (2008). H. pylori-induced apoptosis in human gastric cancer cells mediated via the release of apoptosis-inducing factor from mitochondria. Helicobacter 13 (6), 506–517. doi:10.1111/j.1523-5378.2008.00646.x
PubMed Abstract | CrossRef Full Text | Google Scholar
Backert, S., Tegtmeyer, N., and Fischer, W. (2015). Composition, structure and function of the Helicobacter pylori cag pathogenicity island encoded type IV secretion system. Future Microbiol. 10 (6), 955–965. doi:10.2217/fmb.15.32
PubMed Abstract | CrossRef Full Text | Google Scholar
Bae, S., Lim, J. W., and Kim, H. (2021). β-Carotene inhibits expression of matrix metalloproteinase-10 and invasion in Helicobacter pylori-infected gastric epithelial cells. Molecules 26 (6), 1567. doi:10.3390/molecules26061567
PubMed Abstract | CrossRef Full Text | Google Scholar
Bebb, J. R., Letley, D. P., Thomas, R. J., Aviles, F., Collins, H. M., Watson, S. A., et al. (2003). Helicobacter pylori upregulates matrilysin (MMP-7) in epithelial cells in vivo and in vitro in a Cag dependent manner. Gut 52 (10), 1408–1413. doi:10.1136/gut.52.10.1408
PubMed Abstract | CrossRef Full Text | Google Scholar
Boengler, K., Hilfiker-Kleiner, D., Drexler, H., Heusch, G., and Schulz, R. (2008). The myocardial JAK/STAT pathway: From protection to failure. Pharmacol. Ther. 120 (2), 172–185. doi:10.1016/j.pharmthera.2008.08.002
PubMed Abstract | CrossRef Full Text | Google Scholar
Brandt, S., Kwok, T., Hartig, R., König, W., and Backert, S. (2005). NF-kappaB activation and potentiation of proinflammatory responses by the Helicobacter pylori CagA protein. Proc. Natl. Acad. Sci. U. S. A. 102 (26), 9300–9305. doi:10.1073/pnas.0409873102
PubMed Abstract | CrossRef Full Text | Google Scholar
Cai, B., Cai, J. P., Luo, Y. L., Chen, C., and Zhang, S. (2015). The specific roles of JAK/STAT signaling pathway in sepsis. Inflammation 38 (4), 1599–1608. doi:10.1007/s10753-015-0135-z
PubMed Abstract | CrossRef Full Text | Google Scholar
Chang, H., Chen, D., Ni, B., Zuo, Q., Wang, C., Han, R., et al. (2016). Cortactin mediates apoptosis of gastric epithelial cells induced by VacA protein of Helicobacter pylori. Dig. Dis. Sci. 61 (1), 80–90. doi:10.1007/s10620-015-3836-0
PubMed Abstract | CrossRef Full Text | Google Scholar
Chattopadhyay, R., Bhattacharyya, A., and Crowe, S. E. (2010). Dual regulation by apurinic/apyrimidinic endonuclease-1 inhibits gastric epithelial cell apoptosis during Helicobacter pylori infection. Cancer Res. 70 (7), 2799–2808. doi:10.1158/0008-5472.CAN-09-4136
PubMed Abstract | CrossRef Full Text | Google Scholar
Chen, G. Y., Shu, Y. C., Chuang, D. Y., and Wang, Y. C. (2016). Inflammatory and apoptotic regulatory activity of Tanshinone IIA in Helicobacter pylori-infected cells. Am. J. Chin. Med. 44 (6), 1187–1206. doi:10.1142/S0192415X1650066X
PubMed Abstract | CrossRef Full Text | Google Scholar
Chen, G., Tang, N., Wang, C., Xiao, L., Yu, M., Zhao, L., et al. (2017). TNF-alpha-inducing protein of Helicobacter pylori induces epithelial-mesenchymal transition (EMT) in gastric cancer cells through activation of IL-6/STAT3 signaling pathway. Biochem. Biophys. Res. Commun. 484 (2), 311–317. doi:10.1016/j.bbrc.2017.01.110
PubMed Abstract | CrossRef Full Text | Google Scholar
Chen, Y., Sheppard, D., Dong, X., Hu, X., Chen, M., Chen, R., et al. (2020). H. pylori infection confers resistance to apoptosis via Brd4-dependent BIRC3 eRNA synthesis. Cell. Death Dis. 11 (8), 667. doi:10.1038/s41419-020-02894-z
PubMed Abstract | CrossRef Full Text | Google Scholar
Cheng, A. S., Li, M. S., Kang, W., Cheng, V. Y., Chou, J. L., Lau, S. S., et al. (2013). Helicobacter pylori causes epigenetic dysregulation of FOXD3 to promote gastric carcinogenesis. Gastroenterology 144 (1), 122–133. doi:10.1053/j.gastro.2012.10.002
PubMed Abstract | CrossRef Full Text | Google Scholar
Costa, V. L., Henrique, R., Danielsen, S. A., Duarte-Pereira, S., Eknaes, M., Skotheim, R. I., et al. (2010). Three epigenetic biomarkers, GDF15, TMEFF2, and VIM, accurately predict bladder cancer from DNA-based analyses of urine samples. Clin. Cancer Res. 16 (23), 5842–5851. doi:10.1158/1078-0432.CCR-10-1312
PubMed Abstract | CrossRef Full Text | Google Scholar
Costa, A. M., Ferreira, R. M., Pinto-Ribeiro, I., Sougleri, I. S., Oliveira, M. J., Carreto, L., et al. (2016). Helicobacter pylori activates matrix metalloproteinase 10 in gastric epithelial cells via EGFR and ERK-mediated pathways. J. Infect. Dis. 213 (11), 1767–1776. doi:10.1093/infdis/jiw031
PubMed Abstract | CrossRef Full Text | Google Scholar
Dickson, J. H., Grabowska, A., El-Zaatari, M., Atherton, J., and Watson, S. A. (2006). Helicobacter pylori can induce heparin-binding epidermal growth factor expression via gastrin and its receptor. Cancer Res. 66 (15), 7524–7531. doi:10.1158/0008-5472.CAN-05-3246
PubMed Abstract | CrossRef Full Text | Google Scholar
Ding, S. Z., Smith, M. F., and Goldberg, J. B. (2008). Helicobacter pylori and mitogen-activated protein kinases regulate the cell cycle, proliferation and apoptosis in gastric epithelial cells. J. Gastroenterol. Hepatol. 23, e67–e78. doi:10.1111/j.1440-1746.2007.04912.x
PubMed Abstract | CrossRef Full Text | Google Scholar
Fischer, W., Püls, J., Buhrdorf, R., Gebert, B., Odenbreit, S., and Haas, R. (2001). Systematic mutagenesis of the Helicobacter pylori cag pathogenicity island: Essential genes for CagA translocation in host cells and induction of interleukin-8. Mol. Microbiol. 42 (5), 1337–1348. doi:10.1046/j.1365-2958.2001.02714.x
PubMed Abstract | CrossRef Full Text | Google Scholar
Gao, J. P., Xu, W., Liu, W. T., Yan, M., and Zhu, Z. G. (2018). Tumor heterogeneity of gastric cancer: From the perspective of tumor-initiating cell. World J. Gastroenterol. 24 (24), 2567–2581. doi:10.3748/wjg.v24.i24.2567
PubMed Abstract | CrossRef Full Text | Google Scholar
Gemel, J., Simon, A. R., Patel, D., Xu, Q., Matiukas, A., Veenstra, R. D., et al. (2014). Degradation of a connexin40 mutant linked to atrial fibrillation is accelerated. J. Mol. Cell. Cardiol. 74, 330–339. doi:10.1016/j.yjmcc.2014.06.010
PubMed Abstract | CrossRef Full Text | Google Scholar
Göõz, M., Göõz, P., and Smolka, A. J. (2001). Epithelial and bacterial metalloproteinases and their inhibitors in H. pylori infection of human gastric cells. Am. J. Physiol. Gastrointest. Liver Physiol. 281 (3), G823–G832. doi:10.1152/ajpgi.2001.281.3.G823
PubMed Abstract | CrossRef Full Text | Google Scholar
Guo, Q., Jing, F. J., Qu, H. J., Xu, W., Han, B., Xing, X. M., et al. (2019). Ubenimex reverses MDR in gastric cancer cells by activating caspase-3-mediated apoptosis and suppressing the expression of membrane transport proteins. Biomed. Res. Int. 2019, 4390839. doi:10.1155/2019/4390839
PubMed Abstract | CrossRef Full Text | Google Scholar
Guo, Y., Zhang, T., Shi, Y., Zhang, J., Li, M., Lu, F., et al. (2020a). Helicobacter pylori inhibits GKN1 expression via the CagA/p-ERK/AUF1 pathway. Helicobacter 25 (1), e12665. doi:10.1111/hel.12665
PubMed Abstract | CrossRef Full Text | Google Scholar
Guo, Y., Pan, W. W., Liu, S. B., Shen, Z. F., Xu, Y., and Hu, L. L. (2020b). ERK/MAPK signalling pathway and tumorigenesis. Exp. Ther. Med. 19 (3), 1997–2007. doi:10.3892/etm.2020.8454
PubMed Abstract | CrossRef Full Text | Google Scholar
Hojo, M., Miwa, H., Kikuchi, S., and Sato, N. (2000). Do mucosal defensive agents improve the cure rate when used with dual or triple therapy regimens for eradicating Helicobacter pylori infection? Aliment. Pharmacol. Ther. 14 (2), 193–201. doi:10.1046/j.1365-2036.2000.00692.x
PubMed Abstract | CrossRef Full Text | Google Scholar
Jiang, H., Zhou, Y., Liao, Q., and Ouyang, H. (2014). Helicobacter pylori infection promotes the invasion and metastasis of gastric cancer through increasing the expression of matrix metalloproteinase-1 and matrix metalloproteinase-10. Exp. Ther. Med. 8 (3), 769–774. doi:10.3892/etm.2014.1822
PubMed Abstract | CrossRef Full Text | Google Scholar
Katz, M., Amit, I., and Yarden, Y. (2007). Regulation of MAPKs by growth factors and receptor tyrosine kinases. Biochim. Biophys. Acta 1773 (8), 1161–1176. doi:10.1016/j.bbamcr.2007.01.002
PubMed Abstract | CrossRef Full Text | Google Scholar
Khanna, P., Chua, P. J., Bay, B. H., and Baeg, G. H. (2015). The JAK/STAT signaling cascade in gastric carcinoma (Review). Int. J. Oncol. 47 (5), 1617–1626. doi:10.3892/ijo.2015.3160
PubMed Abstract | CrossRef Full Text | Google Scholar
Kimura, M., Goto, S., Ihara, Y., Wada, A., Yahiro, K., Niidome, T., et al. (2001). Impairment of glutathione metabolism in human gastric epithelial cells treated with vacuolating cytotoxin from Helicobacter pylori. Microb. Pathog. 31 (1), 29–36. doi:10.1006/mpat.2001.0446
PubMed Abstract | CrossRef Full Text | Google Scholar
Koch, K. N., and Muller, A. (2015). Helicobacter pylori activates the TLR2/NLRP3/caspase-1/IL-18 axis to induce regulatory T-cells, establish persistent infection and promote tolerance to allergens. Gut Microbes 6 (6), 382–387. doi:10.1080/19490976.2015.1105427
PubMed Abstract | CrossRef Full Text | Google Scholar
Krueger, S., Hundertmark, T., Kalinski, T., Peitz, U., Wex, T., Malfertheiner, P., et al. (2006). Helicobacter pylori encoding the pathogenicity island activates matrix metalloproteinase 1 in gastric epithelial cells via JNK and ERK. J. Biol. Chem. 281 (5), 2868–2875. doi:10.1074/jbc.M511053200
PubMed Abstract | CrossRef Full Text | Google Scholar
Lan, C. H., Sheng, J. Q., Fang, D. C., Meng, Q. Z., Fan, L. L., and Huang, Z. R. (2010). Involvement of VDAC1 and Bcl-2 family of proteins in VacA-induced cytochrome c release and apoptosis of gastric epithelial carcinoma cells. J. Dig. Dis. 11 (1), 43–49. doi:10.1111/j.1751-2980.2009.00412.x
PubMed Abstract | CrossRef Full Text | Google Scholar
Li, R., Zou, X., Zhu, T., Xu, H., Li, X., and Zhu, L. (2020). Destruction of neutrophil extracellular traps promotes the apoptosis and inhibits the invasion of gastric cancer cells by regulating the expression of bcl-2, Bax and NF-κB. OncoTargets Ther. Vol. 13, 5271–5281. doi:10.2147/OTT.S227331<
CrossRef Full Text | Google Scholar
Lin, K., Taylor, J. R., Wu, T. D., Gutierrez, J., Elliott, J. M., Vernes, J. M., et al. (2011). TMEFF2 is a PDGF-AA binding protein with methylation-associated gene silencing in multiple cancer types including glioma. PLoS One 6 (4), e18608. doi:10.1371/journal.pone.0018608
PubMed Abstract | CrossRef Full Text | Google Scholar
Lin, W. C., Tsai, H. F., Liao, H. J., Tang, C. H., Wu, Y. Y., Hsu, P. I., et al. (2014). Helicobacter pylori sensitizes TNF-related apoptosis-inducing ligand (TRAIL)-mediated apoptosis in human gastric epithelial cells through regulation of FLIP. Cell. Death Dis. 5, e1109. doi:10.1038/cddis.2014.81
PubMed Abstract | CrossRef Full Text | Google Scholar
Liu, F., Zhang, W., Yang, F., Feng, T., Zhou, M., Yu, Y., et al. (2016). Interleukin-6-stimulated progranulin expression contributes to the malignancy of hepatocellular carcinoma cells by activating mTOR signaling. Sci. Rep. 6, 21260. doi:10.1038/srep21260
PubMed Abstract | CrossRef Full Text | Google Scholar
Liu, J. F., Guo, D., Kang, E. M., Wang, Y. S., Gao, X. Z., Cong, H. Y., et al. (2021). Acute and chronic infection of H. pylori caused the difference in apoptosis of gastric epithelial cells. Microb. Pathog. 150, 104717. doi:10.1016/j.micpath.2020.104717
留言 (0)