
記住我


Ehlers Danlos syndrome (EDS) is a heritable disorder of the connective tissue usually inherited as an autosomal dominant trait (1). EDS comprises a genetically heterogeneous group of connective tissue disorders of which the main clinical features are joint hypermobility, skin hyperextensibility, delayed wound healing with atrophic scarring, and generalized connective tissue fragility (2,3). The genetic etiology is not well explained, and there are 100s of variants within the core genes. We have observed an enrichment of EDS cases in a set of gastroparesis patients with idiopathic and diabetic gastroparesis enrolled in a phase II clinical study (VP-VLY-686-2301) (4) and in phase III gastroparesis clinical study (VP-VLY-686-3301). Gastroparesis is a potentially serious medical condition characterized by delayed gastric emptying and symptoms of nausea, vomiting, bloating, fullness after meals, and abdominal pain in the absence of mechanical obstruction (5–7).The overall standardized prevalence of gastroparesis is 267.7 per 100,000 US adult, whereas the prevalence of “definite” gastroparesis is 21.5 per 100,000 (8). The pathophysiology of gastroparesis is complex and probably involves neuromuscular dysfunction and sensory neuropathy, resulting in delayed gastric emptying, nausea, and pain (7).
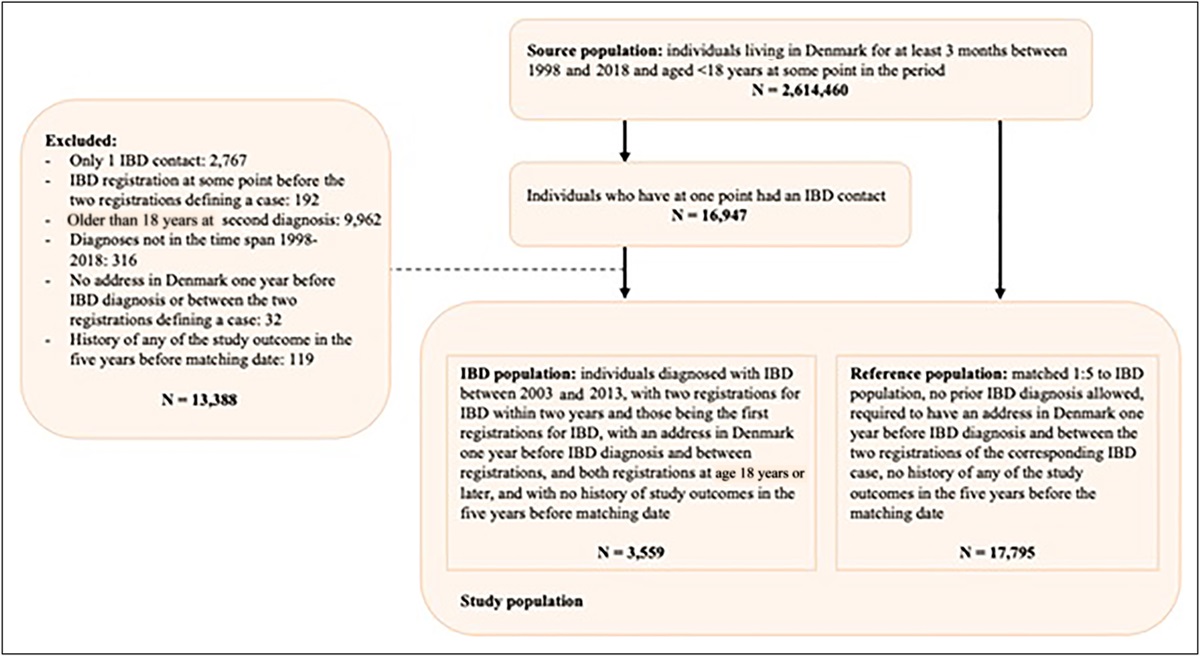
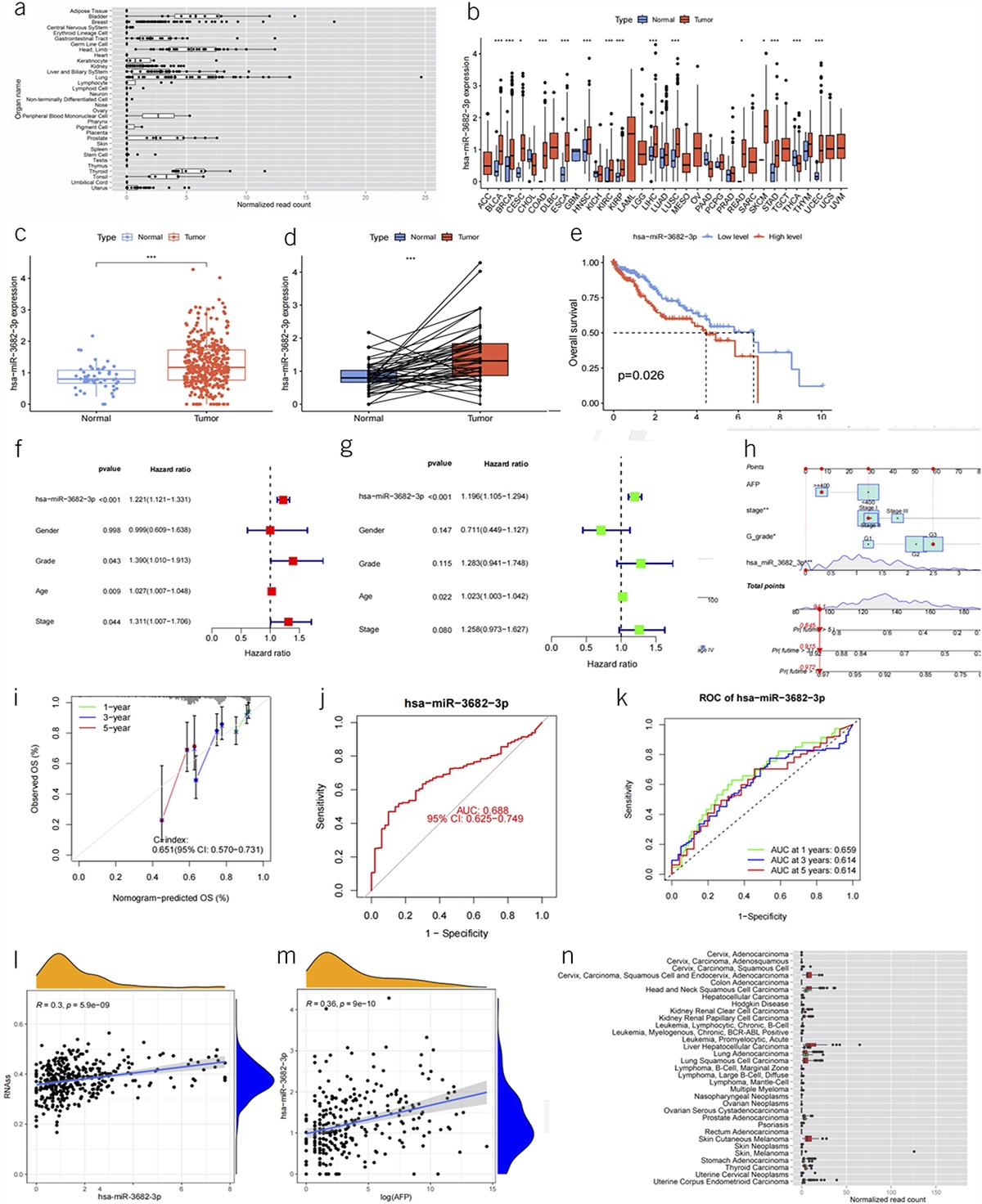
RESULTSIn the combined set of patients enrolled in the phase II and phase III gastroparesis study (686), we report 14 cases of EDS (confirmed by electronic medical records). Assuming the EDS incidence rate of 1 in 5,000 (9), we report a significant enrichment (14/686 vs 1/5,000 OR 104 (confidence interval [CI] 13.7–793.3) P value <0.0001). This is a conservative comparator because other studies have been reporting EDS prevalence at 1 in 20,000 (10). When considering all the screened patients, the enrichment is also significant (18/2065 vs 1/5000 OR 43 [CI 5.8–329.5] P value <0.0002). Importantly, 94% of the patients in this EDS group are female with the idiopathic type of gastroparesis. The average age is 28 years old, and the average body mass index is 27 kg/m2. The detailed demographics and the baseline clinical characteristics of these EDS patients are provided in Table 1. All EDS cases are of White ancestry. All self-reported ancestry was verified by principal component analysis of the genetic sequencing data.
Table 1. - Demographics and the baseline clinical characteristics of the patients with EDS in the gastroparesis cohort Characteristic Baseline value Age, (y) 28.8 (8.78) Male sex—no. (%) 1 (5.6) Ethnicity White—no. (%) 18 (100.0) Disease etiology—no. (%) 18 (100.0) Body mass index, kg/m2 27.3 (6.10) Coexisting condition Nausea severity score—mean (SD) 3.1 (0.87) Nausea-free days—mean (SD) 1.9 (6.13) Hours of nausea—mean (SD) 6.1 (4.85) Frequency of vomiting—mean (SD) 0.6 (0.84) Not able to finish a normal-sized meal—mean (SD) 3.1 (1.45) Feeling excessively full after meals—mean (SD) 2.9 (1.40) Upper abdominal pain—mean (SD) 2.4 (1.22) Bloating—mean (SD) 2.5 (1.26) GCSI—mean (SD) 2.9 (0.91) PAGI-SYM—mean (SD) 2.5 (0.84)Most of the cases are idiopathic female of White ancestry.
EDS, Ehlers Danlos syndrome; GCSI, Gastroparesis Cardinal Symptom Index; PAGI-SYM, Patient Assessment Symptom Severity Index.
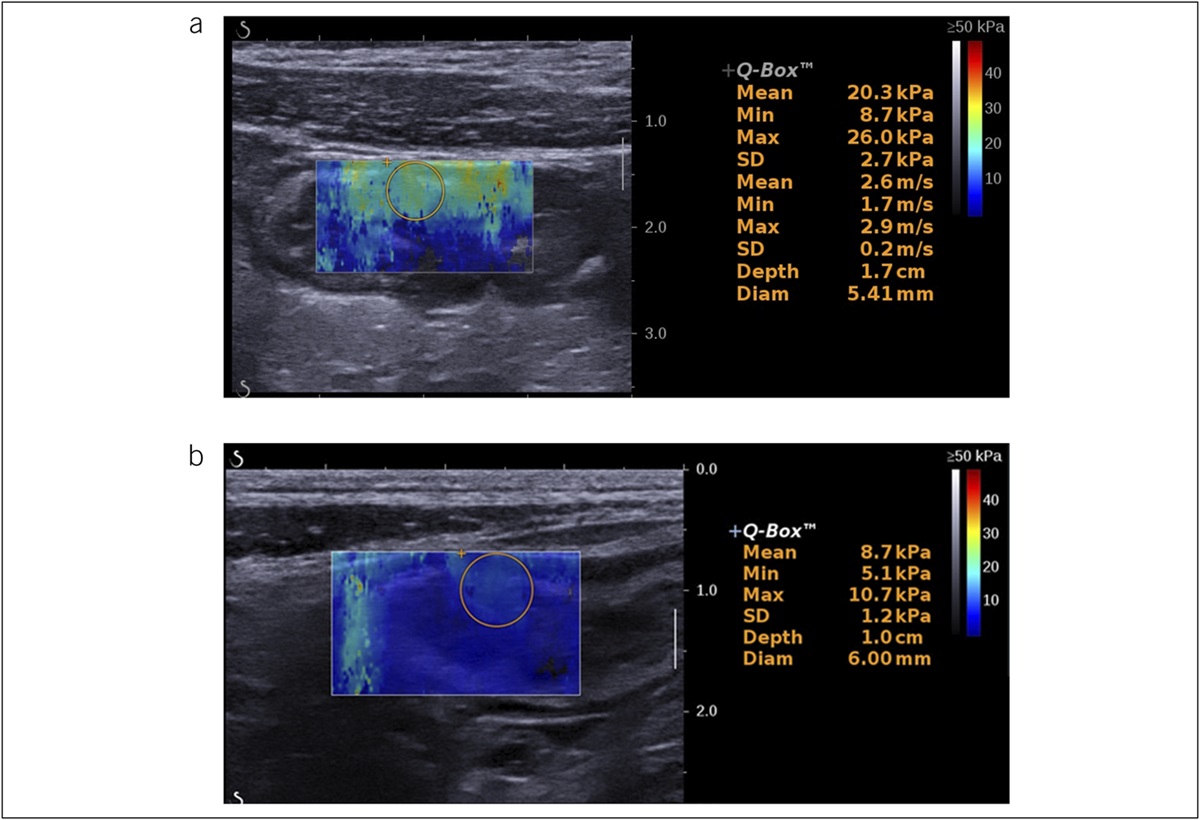
To explore the genetic surrogates of EDS, we focused our analysis on core EDS genes previously associated with EDS: COL1A1, COL1A2, COL3A1, COL5A1, and COL5A2 and ADAMTS2, FKBP14, PLOD1, and TNXB, as reported in LOVD (11). We report all coding/splicing variants in the core genes with a minor allele frequency (MAF) <0.05. Most of our EDS cases for which we have whole-genome sequencing available (n = 13) with exception of 1 have at least 1 such nonsynonymous variant/or splicing variant within the core gene set (Figure 1). We report 2 duplicate variants across this set. The first variant is COL5A1:NM_000093:exon13:c.G1588A:p.G530S (rs61735045). It has a global MAF in gnomAD (12) of 0.03 (ranging from 0.04 non-Finnish to 0.0009 among East Asian). The variant is reported in ClinVar (13) as pathogenic and associated with EDS, classic type and has a high in silico prediction deleteriousness score as defined by the “Combined Annotation Dependent Depletion” score of 24.9. This variant has been previously reported in the literature in association with EDS (1). These findings suggested that heterozygosity for the Gly530Ser substitution is associated with mild ultrastructural abnormalities and variably subtle clinical signs and is disease modifying, while homozygosity for this mutation is associated with classic EDS (1). Furthermore, the variant is significant in association with the analysis of patients with idiopathic vs diabetic gastroparesis. We report 5/128 diabetic carriers vs 27/238 idiopathic, P value <0.007 (EDS gene set). The other variant duplicated in our cohort is the TNXB:NM_019105:exon26:c.A9044G:p.K3015R, with a high Combined Annotation Dependent Depletion score of 23.3. Among the 19 variants (total 21) reported in the 13 patients with EDS, 9 have been reported in association with EDS confirmed in ClinVar (13). In addition, a gene enrichment analysis was performed on the core EDS genes, with MAF <5. In this analysis, we compared idiopathic patients vs nongastroparesis sex, ancestry, and age-matched controls. Interestingly, we report a significantly higher frequency of variants in COL5A1, TNXB, and nominally significant COL1A2 in the patients with idiopathic gastroparesis (Table 2). TNXB enrichment remains significant after Bonferroni correction (P value <0.001). The results of this analysis imply that there may be more cases with underlying EDS and EDS variants than expected by chance in the gastroparesis set, perhaps explaining proportion of the idiopathic etiology.
 Figure 1.:
Figure 1.: Variant gram depicts identified variants in the EDS genes in the set of patients with EDS. All coding/splicing variants in the core EDS genes with a MAF <0.05 are depicted. Altogether, 12 of 13 of the studied patients has at least 1 such nonsynonymous variant/or splicing variant within the core gene set. Two variants occur twice: (COL5A1:p.G530S) and TNXB:p.K3015R. EDS, Ehlers Danlos syndrome; MAF, minor allele frequency.
Table 2. - The results of an enrichment analysis focused on the frequency of EDS gene variants comparing idiopathic gastroparesis patients with diabetic gastroparesis patients Gene name Idiopathic (N = 238) Nongastro (N = 902) P value ADAMTS2 45 200 0.3068 COL1A1 10 58 0.2071 COL1A2 2 31 0.0519 COL3A1 9 48 0.3409 COL5A1 68 187 0.0162 COL5A2 62 255 0.5336 COL5A3 48 150 0.2235 FKBP14 0 10 0.2354 PLOD1 21 58 0.206 TNXB 356 1,067 0.0001 Significant enrichment of variants (MAF a. Reported P values are unadjusted.EDS, Ehlers Danlos syndrome.
aTNXB is significant after conservative Bonferroni correction.
These results suggest a statistically significant enrichment of patients with EDS in this gastroparesis study, with confirmed known and novel variants in the EDS gene set. The enrichment may be suggestive of converging pathways at the heart of etiology or predisposing patients to EDS with gastroparesis. This is in line with the recent reports suggesting that there is a strong correlation between joint hypermobility disorders and gastrointestinal disorders (14). Several other studies documented the evidence of gastric emptying or other transit disorders in patients with EDS (15,16). The composition of the extracellular matrix in which the other components of the gut wall are embedded could ultimately be at the center of the pathogenesis of dysmotility (14). It has been reported that collagen V plays a crucial role in controlling the formation of the heterotypic fibrils and in regulation of their diameter (1). Haploinsufficiency of COL5A1 is a common cause of classic EDS (1). Interestingly, the deficiency of other proteins involved in the formation of collagen fibrils (decorin and thrombospondin-2) may also result in EDS (1). Collagen fibril density was shown to modulate macrophage activation (17). M2-like macrophages were shown to play a key role in the process of the collagen turnover in vivo mice dermis study (18). The EDS enrichment may be a result of turnover of collagen tripeptides and the resulting directional polarization of macrophages resulting in this dual manifestation of EDS in patients with gastroparesis.
These results suggest an enrichment of EDS gene variants specifically in the idiopathic set of patients with gastroparesis. Haploinsufficiency of TNXB was previously described in association with EDS subtypes (19). Furthermore, the extracellular matrix glycoprotein tenascin (TNX) regulates peripheral sensory and motor neurons, and TNX-deficient patients reported increased sensory and motor GI symptoms (20). Another study has reported that TNX-deficient patients have upper gastric dysfunction consistent with those in a mouse model (21). These findings support determination of TNX deficiency in patients reporting connective tissue abnormalities.
The present analysis was focused on enrichment of loss of function variants in the EDS genes. There may be variants of consequence predisposing to risk for EDS outside the core genes tested. A large genome-wide association study based on all the collected data is warranted. These further analyses will potentially help discern any other genetic underpinnings of EDS in a gastroparesis set of patients. It may be the case that there are several individuals who are predisposed because of rare consequential variants, but most of the genetic risk is due to polygenic effects similar to the risk for type II diabetes that is best explained by a polygenic risk score spanning many loci across genes. The identified variants within the reported genomic regions can serve as risk factors for the comorbid disorders as well as one day may inform treatments.
CONFLICTS OF INTERESTGuarantor of the article: Sandra P. Smieszek, PhD.
Specific author contributions: SP Smieszek conducted genetic analysis and wrote the manuscript. JL Carlin led the study. MA Fisher, DS Madonick, CQ Kupersmith, and PD Moszczynski all contributed to the study operations and sample collection. C Xiao conducted the statistical analysis. CM Polymeropoulos, G Birznieks, and MH Polymeropoulos served as mentors.
Financial support: None to report.
Potential competing interests: Authors are employees of Vanda Pharmaceuticals Inc.
REFERENCES 1. Giunta C, Nuytinck L, Raghunath M, et al. Homozygous Gly530Ser substitution in COL5A1 causes mild classical Ehlers-Danlos syndrome. Am J Med Genet 2002;109(4):284–90. 2. Castori M, Dordoni C, Valiante M, et al. Nosology and inheritance pattern(s) of joint hypermobility syndrome and ehlers-Danlos syndrome, hypermobility type: A study of intrafamilial and interfamilial variability in 23 Italian pedigrees. Am J Med Genet Part A 2014;164A(12):3010–20. 3. Malfait F, Hakim AJ, De Paepe A, Grahame R. The genetic basis of the joint hypermobility syndromes. Rheumatology 2006;45(5):502–7. 4. Carlin JL, Lieberman VR, Dahal A, et al. Efficacy and safety of tradipitant in patients with diabetic and idiopathic gastroparesis in a randomized, placebo-controlled trial. Gastroenterology 2021;160(1):76–87. e4. 5. Camilleri M, Parkman HP, Shafi MA, et al. Clinical guideline: Management of gastroparesis. Am J Gastroenterol 2013;108(1):18–37; quiz 38. 6. Atieh J, Sack J, Thomas R, et al. Gastroparesis following immune checkpoint inhibitor therapy: A case series. Dig Dis Sci 2021;66(6):1974–80. 7. Camilleri M, Chedid V, Ford AC, et al. Gastroparesis. Nat Rev Dis Primers 2018;4(1):41. 8. Ye Y, Yin Y, Huh SY, et al. Epidemiology, etiology, and treatment of gastroparesis: Real-world evidence from a large US national claims Database. Gastroenterology 2022;162(1):109–21.e5. 9. Pyeritz RE. Ehlers-Danlos syndrome. N Engl J Med 2000;342(10):730–2. 10. Colombi M, Dordoni C, Cinquina V, et al. A classical Ehlers-Danlos syndrome family with incomplete presentation diagnosed by molecular testing. Eur J Med Genet 2018;61(1):17–20. 11. Fokkema IFAC, Taschner PEM, Schaafsma GCP, et al. LOVD v.2.0: The next generation in gene variant databases. Hum Mutat 2011;32(5):557–63. 12. Karczewski KJ, Francioli LC, Tiao G, et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. bioRxiv 2019:531210. 13. Landrum MJ, Lee JM, Benson M, et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res 2018;46(D1):D1062–7. 14. Mccallum RW, Botrus G, Baker O, et al. Hypermobility syndrome and gastric emptying disorders. Pract Gastroenterol 2016; XL(4). 15. Nelson AD, Mouchli MA, Valentin N, et al. Ehlers Danlos syndrome and gastrointestinal manifestations: A 20-year experience at mayo clinic. Neurogastroenterol Motil 2015;27(11):1657–66. 16. Carbone F, Goelen N, Fikree A, et al. Impact of joint hypermobility syndrome on gastric accommodation and nutrient tolerance in functional dyspepsia. Neurogastroenterol Motil 2021;33(7):e14086. 17. Sapudom J, Mohamed WKE, Garcia-Sabaté A, et al. Collagen fibril Density modulates macrophage activation and cellular functions during tissue repair. Bioengineering (Basel) 2020;7(2):33. 18. Madsen DH, Leonard D, Masedunskas A, et al. M2-like macrophages are responsible for collagen degradation through a mannose receptor-mediated pathway. J Cell Biol 2013;202(6):951–66. 19. Zweers MC, Bristow J, Steijlen PM, et al. Haploinsufficiency of TNXB is associated with hypermobility type of Ehlers-Danlos syndrome. Am J Hum Genet 2003;73(1):214–7. 20. Aktar R, Peiris M, Fikree A, et al. The extracellular matrix glycoprotein tenascin-X regulates peripheral sensory and motor neurones. J Physiol 2018;596(17):4237–51. 21. Aktar R, Peiris M, Fikree A, et al. A novel role for the extracellular matrix glycoprotein-Tenascin-X in gastric function. J Physiol 2019;597(6):1503–15.
留言 (0)