

記住我
Epilepsy is one of the most frequent neurological disorders, with more than 45 million people affected worldwide (Beghi et al., 2019). It is characterized by an enduring predisposition to generate epileptic seizures that result from excessive or hypersynchronous neuronal activity in the brain (Fisher et al., 2014). The etiology of epilepsy is diverse, including structural, genetic, infectious, metabolic, immune as well as unknown causes (Scheffer et al., 2017). Current anti-seizure medications (ASMs) aim to achieve seizure control by suppression of seizure activity. Notwithstanding expansive research, medications that impact epileptogenesis or aim at etiologic factors are not in clinical use. Despite the availability of more than two dozen ASMs, approximately one-third of patients develop drug-resistant epilepsy, i.e., they display ongoing seizures. This leaves them at an increased risk of psychosocial dysfunction, reduced quality of life, and premature death (Loscher et al., 2020). Thus, there is an unmet clinical need to better understand the underlying disease mechanisms and to develop more effective, mechanistically driven therapies.
The last two decades have brought tremendous progress to epilepsy genetics. The advent of next-generation sequencing, e.g., targeted gene panels, whole exome and whole genome sequencing, as well as increasingly powerful bioinformatic tools have led to a surge in gene discovery for monogenic epilepsy syndromes (Moller et al., 2015). Many epilepsy genes encode ion channels, neurotransmitter receptors, solute carriers, synaptic vesicle proteins, transcription factors and proteins involved in metabolic pathways (Howard and Baraban, 2017). The growing knowledge of causative genetic variants spurred the search for personalized therapies that are tailored to a patient’s individual genetic characteristics. In fact, in a small subset of epilepsies, genetic findings have already been translated into effective therapies. Well-established examples are the ketogenic diet for GLUT1 deficiency syndrome (Kass et al., 2016) and vitamin B6 supplementation for ALDH7A1-related epilepsy (Coughlin et al., 2019). However, the discovery of genetic alterations raises the need to interpret their potential functional consequences - a prerequisite for the rational design of individualized treatments. Pathogenic variants within the same gene can produce remarkably different phenotypes. For instance, variants of SCN1A cause a wide spectrum of epilepsies, ranging from mild generalized epilepsy with febrile seizures plus (GEFS+) to severe, intractable epilepsies, such as Dravet syndrome (DS; Catterall et al., 2010; Escayg and Goldin, 2010). Determining functional effects, e.g., loss-of-function (LOF) vs. gain-of-function (GOF), bears immediate consequences for personalized therapy strategies as in SCN2A and GRIN2A/B-related epilepsies (Wolff et al., 2017; Krey et al., 2022). Model systems can help analyze the functional consequences of novel candidate variants in vivo.
In the last decades, epilepsy has been studied in various animal models such as roundworms and zebrafish, but most traditionally in rodents (for comparison see Figure 1; Baraban, 2007; Cunliffe et al., 2015; Johan Arief et al., 2018; Takai et al., 2020; Marshall et al., 2021; Wang and Frankel, 2021). An optimal approach to address genotype–phenotype correlations would be to establish a library of transgenic mice expressing all identified candidate variants. However, such an approach would not only be extremely laborious, but also expensive and time-consuming. Therefore, additional model organisms that allow for large-scale functional studies in a reasonable time frame and in a cost-effective manner are urgently needed. The fruit fly Drosophila melanogaster has emerged as an increasingly attractive model system due to its small size, fast generation time, low maintenance costs and ease of genetic manipulation (Rosch et al., 2019). Recent developments in genome editing techniques have facilitated the generation and characterization of humanized flies, carrying the human epilepsy-causing mutation in the corresponding fly gene (Sun et al., 2012; Schutte et al., 2014; Roemmich et al., 2021). These disease models offer a unique opportunity to unravel the molecular mechanisms underlying genetic epilepsies and to explore potential therapeutic targets. In addition, Drosophila is a well-established model for high-throughput drug screening (Stilwell et al., 2006), eventually providing a rapid and inexpensive platform for the development of novel precision medicine therapies.
Figure 1. Comparison of animal models used in epilepsy research.
1.2. The basics of fruit fliesThe fruit fly Drosophila melanogaster has been widely used as a genetic model organism in biomedical research for more than 100 years. It has greatly advanced our understanding of a broad range of biological processes including genetics, inheritance, embryonic development, learning, and behavior (Jennings, 2011). There are many advantages that make Drosophila an attractive model organism, e.g., the ease of maintenance, cost-effectiveness, fewer ethical restrictions, and the availability of a large and sophisticated genetic toolbox. The genome of Drosophila has been completely sequenced. It comprises around 13,600 protein coding genes that are distributed on four chromosomes (Adams et al., 2000). Approximately 75% of all human disease-related genes have an orthologue in the fly, making it a valuable model organism to study human diseases (Reiter et al., 2001). The short life cycle (10 days at 25°C) of Drosophila is composed of four developmental stages: embryo, larva, pupa, and adult. In addition to the short generation time, the large number of offspring facilitates statistical analysis. The adult fly brain comprises about 100,000 neurons that form discrete circuits that are responsible for complex behaviors such as courtship, sleep, circadian rhythms, learning, and memory (Pandey and Nichols, 2011). Although the anatomic structure of the fly brain differs considerably from that of the human brain, many fundamental functions of neurons, e.g., membrane excitability, voltage-gated ion channels, and neurotransmitter receptors, are highly conserved between the two species (Parker et al., 2011a).
One of the most striking advantages of the fly model is the availability of a large and sophisticated repertoire of genetic tools (reviewed in Hales et al., 2015). For instance, chemical agents, e.g., ethyl methyl sulfonate, or X-ray radiation can be leveraged to introduce random mutations into the genome of the fly. The resulting mutagenized flies can then be examined for a behavioral phenotype of interest. Such forward genetic screens are a suitable method to explore diseases, whose genetic underpinnings have not been elucidated. Conversely, a particular gene of interest can be evaluated for its phenotypic functions by RNA interference (RNAi)-mediated knock-down or gene overexpression. In addition, numerous genetic tools are available to facilitate precise genome editing, including transposable P-elements, homologous recombination, and CRISPR/Cas9. These techniques can be readily used to investigate the disease causality of rare variants found in human patients. A common approach is to knock-out or knock-down the Drosophila orthologue of the respective human gene and to analyze the resulting phenotype. If a phenotypic alteration is observed, wild-type and variant human cDNA are subsequently expressed. The causative nature of a variant may be confirmed if the observed phenotype is ameliorated by the wild-type but not the variant human cDNA (Wangler et al., 2015; Yamaguchi and Yoshida, 2018). All these aspects and techniques positioned Drosophila as a powerful model organism that is studied in a broad range of human diseases including Parkinson’s disease, Alzheimer’s disease, Huntington’s disease, amyotrophic lateral sclerosis, brain tumors, and epilepsy (Jeibmann and Paulus, 2009; Lenz et al., 2013; Prüssing et al., 2013).
2. Drosophila in epilepsy research 2.1. The class of bang-sensitive mutantsDrosophila has been used in epilepsy research since the discovery of the group of bang-sensitive mutants more than 50 years ago (Benzer, 1971). These flies typically respond with seizure-like behavior and paralysis to a mechanical shock (termed “bang”), such as a tap of the culture vial on the bench top or a brief vortex mixing (Song and Tanouye, 2008; Parker et al., 2011a; Burg and Wu, 2012). This complex behavioral phenotype can be divided into six distinguishable stages: (1) a shock-induced “initial seizure” that lasts several seconds and that is characterized by extensive wing flapping, leg shaking, abdominal contractions, and proboscis extensions; (2) a post seizure “paralysis” where the flies are completely immobile and do not respond to mechanical stimulation; (3) a “tonic–clonic phase” where the paralytic behavior is interrupted by multiple bouts of clonus-like activity (only observed in a fraction of bang-sensitive flies); (4) a “recovery seizure” that resembles the initial seizure and clonus-like activity; (5) a “refractory period” during which the flies exhibit normal behavior but are resistant to further seizure induction; and (6) a complete “recovery” where the flies re-acquire their bang-sensitivity. In addition to mechanical stimulation, the seizure-like phenotype can also be induced by high frequency electrical stimulation directly delivered to the fly brain. Each genotype has a specific seizure threshold at which seizure-like behavior occurs. Even wild-type flies will display seizure-like behavior if the voltage is high enough. However, the seizure threshold of bang-sensitive mutants is significantly lower than that of wild-type flies (Kuebler and Tanouye, 2000; Howlett and Tanouye, 2009). The bang-sensitive phenotype can be attenuated by treatment with several ASMs, e.g., valproate, phenytoin, gabapentin, and potassium bromide (Kuebler and Tanouye, 2002; Reynolds et al., 2003; Tan et al., 2004). In addition, mutations in specific genes can (partially) revert the behavioral phenotype of bang-sensitive mutants. Prominent examples of seizure-suppressor mutations include alleles of Shaker (Sh) and shaking B (shakB), which encode a potassium channel and a gap junction protein, respectively (Song and Tanouye, 2008). The identification of such seizure-suppressor mutations is challenging in humans but a well-established practice in Drosophila.
Since human epilepsy syndromes are often caused by mutations in genes encoding voltage-gated sodium channels (Escayg and Goldin, 2010; Johannesen et al., 2019), it is not surprising that a particular member of the bang-sensitive mutant class, i.e., bang senseless (parabss1), was found to carry a gain-of-function mutation in the paralytic (para) gene (Parker et al., 2011b). It encodes the only Drosophila voltage-gated sodium channel α-subunit and is orthologous to SCN1A to SCN5A and SCN7A to SCN11A in humans (Takai et al., 2020). The phenotype of parabss1 is caused by a single amino acid substitution at position 1,699 (i.e., L1699F), which is located within the “paddle motif” of homology domain IV. Electrophysiology experiments in Xenopus oocytes showed that the mutation leads to a shift in voltage dependence of fast inactivation to more positive potentials (Parker et al., 2011b). This is consistent with the hypothesis that the voltage sensor paddle of homology domain IV is crucial for channel inactivation (Alabi et al., 2007; Bosmans et al., 2008). Thus, the L1699F variant renders neurons expressing parabss1 more excitable and flies more prone to produce seizures (Parker et al., 2011b). The parabss1 mutant is characterized by the most severe phenotype and the lowest seizure threshold of all bang-sensitive mutants. In addition, the phenotype of parabss1 is the most difficult to suppress by ASMs or seizure-suppressor mutations, making it a suitable model for intractable epilepsy (Kroll et al., 2015a).
Another member of the bang-sensitive mutant class has been shown to carry a frame-shift mutation in the easily shocked (eas) gene, which encodes ethanolamine kinase, an enzyme involved in the synthesis of the membrane lipid phosphatidylethanolamine. The mutation results in a truncated protein that lacks enzymatic activity. It has been suggested that the bang-sensitive phenotype is caused by an altered membrane phospholipid composition (Pavlidis et al., 1994). Furthermore, there are several bang-sensitive mutants with impaired mitochondrial function. The affected genes include technical knockout (tko), stress-sensitive B (sesB), and knockdown (kdn), which encode a mitochondrial riboprotein, an ATP translocase, and citrate synthase, respectively (Royden et al., 1987; Zhang et al., 1999; Fergestad et al., 2006). ATP levels in these mutants are decreased, nurturing the hypothesis that metabolic perturbations may alter neuronal activity and increase seizure susceptibility (Fergestad et al., 2006). The underlying mechanism could be the impaired ability to maintain ionic gradients across the plasma membrane since the Na+/K+ ATPase is a large consumer of neuronal ATP. This is consistent with the observation that a specific mutation in the Na+/K+ ATPase α-subunit gene (i.e., the 2206 mutation) also results in a mild bang-sensitive phenotype (Schubiger et al., 1994; Pavlidis and Tanouye, 1995).
2.2. Other Drosophila mutants in epilepsy researchBesides bang-sensitive mutants, several other classes of Drosophila mutants have been used in epilepsy research. A prominent example is the group of temperature-sensitive paralytic mutants, which typically exhibit behavioral paralysis at elevated temperatures. A well-known member of this group is the maleless (mle) allele called no-action potential temperature-sensitive (mlenapts; Song and Tanouye, 2008). Interestingly, adult mlenapts flies display a reduction of voltage-gated sodium channels in their brains (Kauvar, 1982; Reenan et al., 2000). Importantly, mlenapts has been shown to suppress seizures in bang-sensitive mutants in homozygous double-mutant condition (Kuebler et al., 2001). Another example of a temperature-sensitive paralytic mutant is paraST76, which carries a loss-of-function mutation in the Drosophila sodium channel gene para (Parker et al., 2011a). Like mlenapts, this variant acts as a seizure suppressor for bang-sensitive mutants (Kuebler et al., 2001).
Another class of Drosophila mutants that have been used in epilepsy research are the so-called leg-shaking mutants. Prominent examples of this group include alleles of shaker (sh) and ether a go-go (eag), which encode different types of potassium channel subunits. Mutants of these genes display rapid leg-shaking in response to ether anesthesia, have heightened metabolic rates, and reduced life spans (Wang et al., 2000). Although the leg-shaking phenotype shares some similarities with the bang-sensitive phenotype, several mutants of these genes have been shown to have higher seizure thresholds than wild-type flies. This is in clear contrast to the bang-sensitive mutants, which typically display reduced seizure thresholds (Kuebler et al., 2001).
2.3. Modelling human SCN1A-related epilepsies in DrosophilaMutations in the human SCN1A gene are known to cause GEFS+ and DS (Catterall et al., 2010), which are both autosomal dominant disorders. GEFS+ is a familial epilepsy syndrome that comprises a wide spectrum of clinical phenotypes. Affected individuals within GEFS+ families most commonly show febrile seizures that may persist beyond the age of 6 years. Other seizure types, such as absence, myoclonic, atonic, or focal seizures, may also be observed (Scheffer and Berkovic, 1997; Singh et al., 1999). Seizures are usually well controlled with ASMs (Catterall et al., 2010). In contrast, DS is a severe form of genetic epilepsy characterized by prolonged, febrile and afebrile, hemiclonic or generalized clonic seizures with onset in the first year of life. Over time, other seizure types appear including myoclonic and atypical absence seizures. Seizures are often refractory to treatment and patients display several other clinical features, such as cognitive, behavioral, and motor impairments (Mei et al., 2019; Zuberi et al., 2022). To investigate the underlying disease mechanisms of these both disorders, several GEFS+ and DS mutations have been knocked into the orthologous Drosophila gene para at corresponding positions (Sun et al., 2012; Schutte et al., 2014; Roemmich et al., 2021). The first variant to be explored was the GEFS+ mutation K1270T in the transmembrane segment 2 of homology domain III (Sun et al., 2012). Since para is located on the X chromosome, homozygous female (paraGEFS+/paraGEFS+) and hemizygous male (paraGEFS+/Y) flies were separately assessed. Mutant flies exhibited heat-induced seizures after immersion of the vials containing the flies in a 40°C water bath, regardless of sex. Seizure activity ceased abruptly after removal of the vials from the water bath, i.e., the flies remained motionless and unresponsive for varying time periods. The observed phenotype is reminiscent of the K1270T phenotype in humans, which features febrile seizures. Since GEFS+ is an autosomal-dominant disorder in humans (i.e., patients are heterozygous), heterozygous female flies (paraGEFS+/control) were also assessed. Heterozygous flies exhibited a significantly reduced seizure probability and a delayed seizure onset time compared to homozygous paraGEFS+ flies. Also, the seizure activity could start and stop more than once. The cessation of movement upon removal from the water bath was only observed in 46% of the seizing flies. The authors concluded that the heat-induced seizure phenotype of K1270T flies is semi-dominant with variable penetrance. Electrophysiological recordings from GABAergic interneurons of adult mutant flies revealed that the deactivation threshold for persistent currents shifts to a more negative voltage when the temperature is increased. This results in prolonged depolarizations in GABAergic neurons, which causes a decrease in inhibitory activity due to reduced neuronal firing. Thus, it was suggested that the temperature-sensitive seizure phenotype is caused by an overall loss of inhibition (Sun et al., 2012).
A DS-associated SCN1A variant that has been explored in Drosophila is S1231R. The mutation is located in the transmembrane segment 1 of homology domain III (Schutte et al., 2014). S1231R flies (paraDS) also exhibited a heat-induced seizure phenotype with a significantly increased heat-sensitivity compared with paraGEFS+ flies. This aligns with the more severe phenotype of DS observed in humans. However, paraDS flies did not show a cessation of movement upon removal from the water bath. Electrophysiological studies of GABAergic interneurons revealed a constitutional and heat-induced reduction in sodium currents, which resulted in a reduction of repetitive neuronal firing. Besides, the effects of the serotonin precursor 5-hydroxytryptophan (5-HTP) on the seizure phenotypes of paraGEFS+ and paraDS flies were investigated. Remarkably, treatment of paraGEFS+ flies with 5-HTP caused an increase in seizure probability at high temperatures, whereas treatment of paraDS flies resulted in a suppression of seizures. Electrophysiological studies showed that the treatment did not affect sodium channel properties, but the reduced burst firing frequency was partially rescued. These observations are consistent with the anti-seizure effects of the serotonin releaser fenfluramine in patients with DS (Lagae et al., 2019; Nabbout et al., 2020).
Another study focused on two different SCN1A variants that occur at the same position in segment 4 of homology domain IV (Roemmich et al., 2021). While the R1648H (R-H) variant is associated with GEFS+, the R1648C (R-C) variant causes DS. To address the question why different forms of epilepsy arise from mutations at the same amino acid position, Drosophila lines carrying either the R-H or R-C mutation were generated by CRISPR/Cas9 gene editing. Flies homozygous for R-H and R-C were lethal, whereas flies heterozygous for these mutations showed spontaneous and heat-induced seizures as well as reduced life spans. Notably, the seizure activity was largely similar between the two mutant strains. Electrophysiological recordings from adult GABAergic neurons showed that both mutations cause sustained depolarizations and reduced firing rates that are exacerbated at elevated temperature. Furthermore, a hyperpolarized deactivation threshold in paraR-C and paraR-H sodium currents was observed, which was present at both room temperature and elevated temperature. Taken together, the similar results from behavioral and electrophysiological studies indicate that the different phenotypes observed in humans may be mainly due to differences in genetic background rather than distinct changes in sodium channel function (Roemmich et al., 2021).
3. Comparison of human and Drosophila epilepsy genes 3.1. Determination of orthologous genesTo study genetic epilepsies in animal models, a robust conservation rate of human disease-associated genes in the respective model organism is a prerequisite. We aimed to compare the orthology of human and Drosophila epilepsy genes. Human genes were defined as “epilepsy genes” if they were reported in the review article “Epilepsy-associated genes” by Wang et al. (2017b). To also account for genes identified after the publication date, we also included genes recorded as developmental and epileptic encephalopathy (DEE)-causative genes in Online Mendelian Inheritance in Man (OMIM; https://omim.org/entry/308350) by September 2022 (see full list in Supplementary Table 1). Drosophila orthologues of human genes and human orthologues of Drosophila genes were defined as genes which had the best score listed on the Drosophila online database platform FlyBase as described previously (Takai et al., 2020). In our paradigm, a minimum score of 6 out 15 possible homology assignments was used as cutoff to define orthologous genes. If two or more genes had identical scores, all genes were counted as orthologues. To define “epilepsy-related” genes in Drosophila, a search on FlyBase was conducted using the keyword “epilepsy” and the filters “D. melanogaster” and “gene”. All genes listed by September 2022 were considered as “epilepsy-related” genes in Drosophila (see full list in Supplementary Table 1).
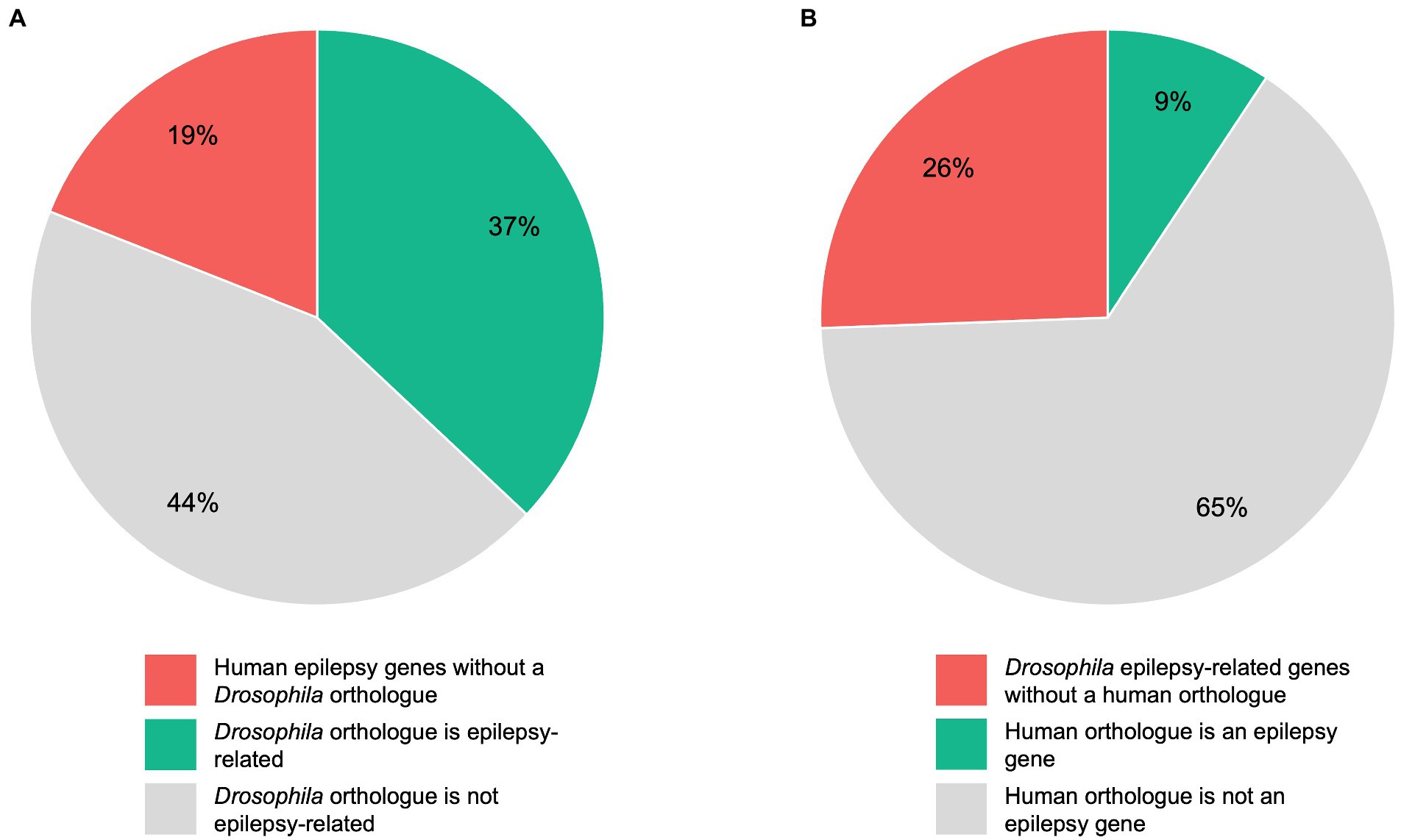
3.2. Conservation rate of human epilepsy genes in DrosophilaTo estimate the conservation rate of human epilepsy genes in Drosophila, we defined a total of 145 human genes as “epilepsy genes” as described above. Although 75% of human disease-related genes are conserved in flies, we found that this percentage is even higher for genes linked to epilepsy. In detail, we found that 117 of the 145 (81%) human epilepsy genes have an orthologous gene in Drosophila, whereas 28 (19%) have no clear orthologue in flies (Figure 2A). To determine which of the orthologous genes are already epilepsy-related in Drosophila, we conducted a search on FlyBase. A total of 344 Drosophila genes matched our search criteria and were defined as “epilepsy-related” genes. It should be noted that not all of these genes can be defined as “epilepsy-causing” genes as some of the genes are associated with seizure-suppressor mutations, e.g., topoisomerase 1 (top1; Song et al., 2008). According to our search, 145 genes have been linked to epilepsy in humans. Out of these 145 genes, 53 (37%) genes were found to have an epilepsy-related orthologue in Drosophila, whereas 64 (44%) genes were found to have a Drosophila orthologue that is not epilepsy-related (Figure 2A). Conversely, we aimed to determine how many of the epilepsy-related genes in Drosophila have a human orthologue. In Drosophila, there are 344 epilepsy-related genes. Out of these 344 genes, 256 (74%) genes were found to have an orthologous gene in humans, whereas no human orthologues were found for 88 (26%) genes (Figure 2B). Of the 344 epilepsy-related Drosophila genes, 32 (9%) genes were found to have a human epilepsy gene as an orthologue, whereas 224 (65%) genes were found to have a human orthologue that is not an epilepsy gene (Figure 2B). A list of epilepsy-related Drosophila genes and their human orthologues including OMIM numbers (if the human orthologue is an epilepsy gene) can be found in Supplementary Table 2.
Figure 2. Comparison of human and Drosophila epilepsy genes. A total of 145 human genes were defined as “epilepsy genes”, whereas 344 Drosophila genes were defined as “epilepsy-related” genes as described under “Determination of orthologous genes”. Pie chart (A) shows the 145 human epilepsy genes divided into those genes without a Drosophila orthologue, genes whose Drosophila orthologue is epilepsy-related, and genes whose Drosophila orthologue is not epilepsy-related. Pie chart (B) shows the 344 epilepsy-related Drosophila genes divided into those genes without a human orthologue, genes whose human orthologue is an epilepsy gene, and genes whose human orthologue is not an epilepsy gene.
3.3. Prediction of novel human epilepsy genes using the Drosophila modelSince there are 224 epilepsy-related Drosophila genes whose human orthologues are not defined as epilepsy genes, it is conceivable that some of these human orthologues could be involved in human epilepsy. Hence, we screened for human genes that are orthologous to more than one of these 224 epilepsy-related Drosophila genes since a higher number of epilepsy-related Drosophila orthologues may indicate a higher risk of the respective human gene to be associated with epilepsy. Indeed, we found several human genes that are orthologues to more than one of these 224 epilepsy-related Drosophila genes (see full list in Supplementary Table 3). For instance, the human SLC2A8 gene is orthologous to 16 of these 224 epilepsy-related Drosophila genes. SLC2A8 encodes a facilitative glucose transporter (Adastra et al., 2012) that is expressed in a wide variety of tissues, such as testis, brain, liver, heart, and fat (Carayannopoulos et al., 2000; Ibberson et al., 2000). SLC2A8 has not been previously associated with epilepsy. Importantly, it should be noted that mutations in another member of the SLC2 family, i.e., SLC2A1 (encoding the glucose transporter type 1), are associated with a variety of distinct epilepsy syndromes, e.g., early onset absence epilepsy, childhood absence epilepsy, and myoclonic-atonic epilepsy (Hildebrand et al., 2014; Koch and Weber, 2019). However, there are also examples where an association with human epilepsy seems rather unlikely. For instance, the human CPB1 gene, which is orthologous to 8 of these 244 epilepsy-related genes in Drosophila, encodes the pancreatic secretory enzyme carboxypeptidase B1. Notably, variants of this gene have been implicated in an increased risk of developing pancreatic cancer (Kawamoto et al., 2022). A link to epilepsy has not been reported yet. Nonetheless, these Drosophila genes represent a valuable resource that may help to identify novel human epilepsy genes. Therefore, further analysis of these genes might be a reasonable approach.
4. Drosophila genetics and tools to investigate seizures in flies 4.1. Genetic tools available in DrosophilaDrosophila as a model organism offers one of the most extensive genetic toolkits available (Senturk and Bellen, 2018) and a nervous system that allows to investigate complex behavior (Ecovoiu et al., 2022). A high conservation rate for human disease-associated genes of ~75% makes it an attractive model to study genetic variants found in humans (Takai et al., 2020). In epilepsy, many severe syndromes are caused by monogenic variants. Information regarding the functional consequences of these variants on the epileptic phenotype is often lacking. As a consequence, the decision on adequate medication can be difficult (Guerrini et al., 2021). Functional analysis of individual disease gene variants in a highly accessible model system is therefore desirable. Drosophila offers this possibility in compliance with reasonable labor input and cost. The existing genetic toolkit for Drosophila allows in-depth studies of genetic networks and protein function and stock centers all over the world provide many of the necessary fly strains (Bloomington Drosophila Stock Center at https://bdsc.indiana.edu; Kyoto Stock Center at http://www.dgrc.kit.ac.jp; Vienna Stock Center at https://stockcenter.vdrc.at). Generally, two approaches can be taken when assessing disease-related variants in Drosophila: Forward-genetics and reverse-genetics (Senturk and Bellen, 2018). In a forward genetic screen, the effects of randomly generated genetic alterations on a specific phenotype are monitored. Unbiased screens for mutations, which either enhance or suppress a certain phenotype (enhancer/suppressor screens) provide a basis for understanding the biological role and interconnection of genes (St Johnston, 2002). They can be used to identify novel therapeutic targets, which might otherwise be overlooked. Screens with chemical agents or transposon-based mutagenesis, which randomly alter parts of the genome, have led to the discovery of several genes associated with seizure-like behavior in flies. Furthermore, screens with these seizure-sensitive flies also enabled the identification of seizure-suppressing variants (Parker et al., 2011a). Reverse genetics seeks to analyze how a phenotype is controlled by a specific genetic sequence. As flies have orthologues for many disease-related human genes, variants first discovered in patients can be investigated for functional aspects in Drosophila. Several techniques for introducing these patient-derived variants into the flies are available.
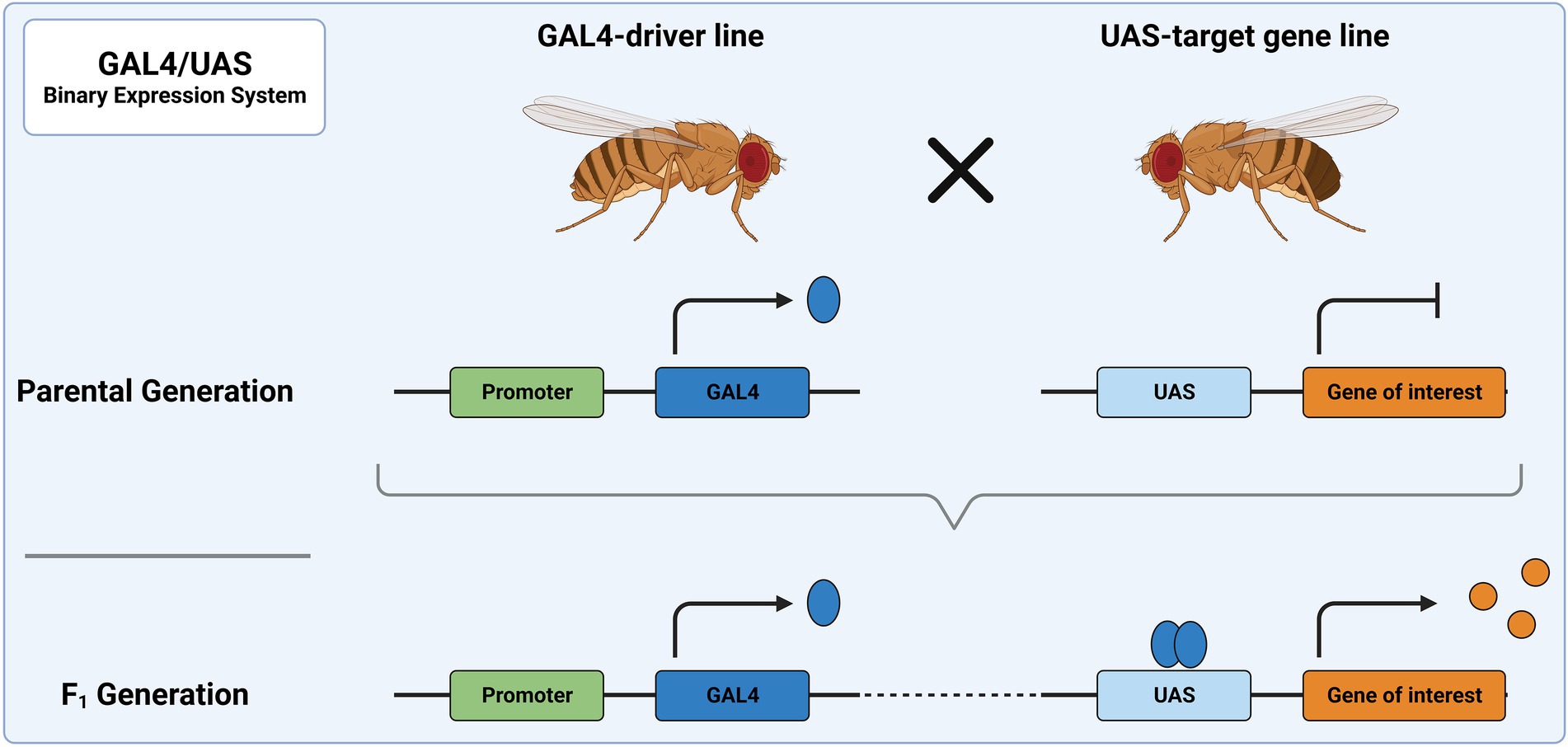
4.1.1. Binary expression systemsAmong the primary genetic tools used in Drosophila are binary expression systems, e.g., the GAL4/UAS, LexA/LexAop or Q-System (Brand and Perrimon, 1993; Lai and Lee, 2006; Riabinina and Potter, 2016). As the name implies, two components are needed to induce gene expression: a driver of gene expression and a sequence whose expression is controlled by the driver. The driver usually consists of a transcription activator, which is expressed under a tissue-specific promoter. This activator then binds a specific genetic sequence and drives expression of a gene linked to this sequence. In the most frequently used binary expression system, the GAL4/UAS system, expression of a UAS-transgene is achieved through the exogenous (yeast) transcription activator protein GAL4 (Figure 3). The UAS-line harbors a gene/sequence of choice downstream of GAL4 binding sites, called upstream activating sequence (UAS). The UAS-controlled sequence is not expressed in the absence of GAL4. Expression of the UAS-controlled sequence is achieved by crossing a GAL4-driver line with a UAS-line. In the progeny, GAL4 will bind to UAS and facilitate expression of the target sequence downstream of UAS. The GAL4-driver lines were generated as random integration enhancer traps. This way, expression of GAL4 is controlled by an endogenous promoter, which defines the expression domain and allows restricting expression to a region of interest. To date, a plethora of characterized driver lines (> 10,000) are available at aforementioned stock centers and many transgene carrying UAS-lines are available there as well. Some of the most frequently used GAL4-driver lines can be found on FlyBase. Drivers commonly used in the study of the nervous system are, for example, elav-GAL4 or nsyb-GAL4, which allow for pan-neuronal expression of GAL4. Further examples include VGlut-GAL4 and ChAT-GAL4 for confining GAL4 expression to glutamatergic and cholinergic neurons, respectively. The expression of GAL4 follows the expression pattern of the associated gene, which might vary during development of the fly. This should be considered when designing experiments.
Figure 3. The GAL4/UAS system represents the most widely used binary expression system. In the parental generation, one fly carries GAL4 under a specific promoter and the other a genetic sequence downstream of UAS. When these flies are crossed, the progeny will express GAL4 in all tissues, in which the promoter is utilized. There, it acts as a transcription factor and binds to UAS, leading to expression of the downstream sequence.
Binary expression systems can be used, for example, to overexpress cDNA from a human disease-related gene or the corresponding Drosophila orthologue (Jenett et al., 2012; Perkins et al., 2015). This cDNA can either be wild-type or a disease-associated variant. If a disease phenotype is observed after expression of the disease-associated variant, but not after expression of the wild-type, the variant is likely a toxic GOF (Prüssing et al., 2013). This approach has been used to classify parabss1 as a GOF variant (Parker et al., 2011b) or to replicate a severe DEE syndrome by overexpression of RhoBTB (Straub et al., 2018). Another possible application of binary expression systems is the induction of RNAi. If a LOF variant is the hypothesis for the disease, RNAi can be used to model the loss of protein function. Expression of inverted repeats (ir) or short hairpins (sh) causes formation of double stranded RNA (dsRNA). If the dsRNA is complementary to endogenous mRNA, this leads to the degradation of target mRNA and a decrease in protein levels (Kaya-Copur and Schnorrer, 2016; Heigwer et al., 2018). The strength of RNAi is influenced by several factors including the levels of interfering RNA, which in turn depends on the activity of the GAL4/UAS system.
To understand the effect of a variant on the phenotype, a rescue experiment can be performed (Ecovoiu et al., 2022). Here, a disease-related phenotype is alleviated by reintroducing a functional allele. For instance, in the case of an epilepsy-related variant in the gene eas, expressing wild-type eas alleviated the seizure phenotype (Kroll and Tanouye, 2013). Besides already established or newly found epilepsy-related variants in genes, RNAi or deficiency lines can be used as a background for rescue experiments. If a wild-type allele rescues the disease phenotype and a variant does not, this provides compelling evidence for a LOF variant.
Though the GAL4/UAS system is a powerful tool, it requires several considerations. The timing of gene expression is not variable and expression strength is generally elevated at higher temperatures, as GAL4 activity increases. To allow for more precise temporal control over gene expression, inducible GAL4 systems can be employed (Supplementary Figure 1). Additional expression of GAL80 can prevent target gene transcription by inhibition of GAL4 (Suster et al., 2004). Moreover, the temperature-sensitive variant GAL80ts is only active at lower temperatures (Ma and Ptashne, 1987; McGuire et al., 2004; del Valle Rodriguez et al., 2011). This way, temporal control of GAL4 activity is achieved by shifting flies to higher temperatures to inactivate GAL80ts. Instead of depending on temperature shifts, the GeneSwitch-GAL4 system depends on the presence of the steroid mifepristone to activate GAL4, which can be added to the standard feeding medium of flies (Osterwalder et al., 2001; del Valle Rodriguez et al., 2011). To allow for better spatial control of GAL4 expression, the split-GAL4 system can be utilized. Here, two different promoters drive expression of a truncated GAL4. Only in cells where both promoters are active, the complementary GAL4 peptides assemble a functional GAL4 (Luan et al., 2020). Even with more specific tools available, these versions of binary expression systems do not fully capture expression profiles of genes of interest, though their easy accessibility makes them useful in initial investigations and for large screens.
4.1.2. Increasing precision of expressionThe phenotypic outcome of a given epilepsy-linked variant might be dependent on gene dosage or timing. Here, the mentioned binary expression systems might fall short. To allow better control over expression levels, an endogenous promoter can be hijacked. The transgene can then be expressed in the same pattern of a gene of interest. This can be achieved by utilizing a Minos-mediated integration cassette (MiMIC; Venken et al., 2011). The transposable element Minos randomly integrates in the genome and harbors a pair of attP landing sites. Via the bacteriophage-derived ΦC31 integrase, recombination mediated cassette exchange (RMCE) can be performed. Ultimately, this creates a system with defined integration sites in the Drosophila genome, in which a desired genetic sequence can be integrated. Different alterations can be achieved based on the position of the insertion. To drive gene expression, the RMCE site can be placed under an endogenous promoter. By targeting an intron, it can be used to tag a protein with a fluorescent protein. As this system is based on random integrations, the efficiency in creating new lines diminishes with each new line generated.
Besides Minos, CRISPR-based strategies (CRIMIC) can also be used to introduce RMCE sites into the genome. Here, integration can be targeted to any position in the genetic sequence accessible to CRISPR/Cas9 modifications. For introns accessible with either method, T2A-GAL4 (“Trojan exon”) can be used to express GAL4 under a promoter of the gene, whose own expression is simultaneously suspended (Diao et al., 2015; Lee et al., 2018). GAL4 can then drive the expression of any UAS construct, while its own expression is controlled by the promoter of the disabled gene. Using this system, different variants can be rapidly screened for their ability to restore protein function, while following endogenous expression patterns. Mutations in the human gene OGDHL cause a neurodevelopmental spectrum disease featuring epilepsy. Using T2A-GAL4, gene function could be restored by wild-type expression, but only partially by patient variants introduced into the Drosophila orthologue dOgdh (Yap et al., 2021). In a similar approach, LOF variants in TIAM1, which are associated with seizures, were introduced into Drosophila and screened for their ability to rescue a loss of the Drosophila orthologue sif (Lu et al., 2022). The lower rescuing capacity of patient variants corroborated a LOF effect. As not all genes are suitable targets for CRMIC-based integration of a trojan exon, the gene can also be replaced completely by a GAL4-expressing sequence (Kanca et al., 2022). Large libraries of flies with integration sites in different genes provide broad access to the Drosophila genome (Venken et al., 2011; Nagarkar-Jaiswal et al., 2015; Lee et al., 2018).
4.1.3. Direct gene modificationsThe most detailed analysis of protein function can be achieved by directly altering the gene itself in the fly genome. In case a human epilepsy-linked mutation is located in a conserved region, the fly gene can be mutated at the respective position to create a humanized variant. Ends-out homologous recombination allows the replacement of the wild-type gene by a mutated variant thereof (Rong and Golic, 2000). Using this technique, patient variants associated with GEFS+ (Sun et al., 2012) or DS (Schutte et al., 2014) have been introduced into the fly genome. To achieve precise genome modifications more efficiently, the CRISPR/Cas9 system has become a widely used tool for genome editing (Jinek et al., 2012; Gratz et al., 2015). In flies, it has been used to introduce patient-specific variants into the gene para, which result in either GEFS+ or DS (Roemmich et al., 2021). This allows to study patient-derived variants in a comparable genetic background and to assess, which physiological alterations might be caused by the variants.
Through these methods, genomic modification on different scales becomes feasible for many conserved genes in Drosophila. Libraries of fly strains generated by these methods provide researchers access to nearly every gene in Drosophila, while direct gene editing remains a possibility to investigate single genes in greater detail.
4.2. Techniques for induction and measurement of seizure-like behaviorThere are several experimental options to induce seizure-like behaviors in Drosophila. For example, flies carrying specific genetic variants are susceptible to mechanical or heat stressors and display seizure-like behavior or paralysis upon exposure to these stressors. The class of bang-sensitive mutants respond to mechanical disturbances (Lee and Wu, 2002) while flies carrying mutations in other genes react to elevated temperatures with seizure-like behavior. Interestingly, some mutant variants only respond to mechanical disturbances or elevated temperature with seizure-like behavior, while others respond to both (Burg and Wu, 2012).
In contrast to rodent models, chemical seizure induction has only rarely been used in Drosophila. Treatment with the proconvulsant picrotoxin (PTX) has been shown to induce seizures in larvae (Stilwell et al., 2006). In addition, flies fed with pentylenetetrazole (PTZ) for 7 days displayed a decrease in climbing speed and transcriptomic alterations, which were similar to those reported in human epileptogenesis. Furthermore, an increase in climbing speed was detected 7 days after withdrawal of PTZ (Mohammad et al., 2009). Of note, treatment with ASMs after PTZ withdrawal alleviated the increased climbing speed and normalized PTZ withdrawal-induced transcriptomic changes (Singh et al., 2011).
4.2.1. Techniques for behavioral seizure assays in DrosophilaTo induce a bang-sensitive phenotype, it has become the standard to use a laboratory “vortex mixer” to shake flies in an empty vial for ~10 s (Kuebler and Tanouye, 2000). If flies have the bang-sensitive phenotype, they display seizure-like behavior and become paralyzed, while wild-type flies remain unaffected. The time until affected flies regain a standing position or mobility correlates with the severity of the phenotype. Administration of ASMs has been shown to reduce the severity of the phenotype in some bang-sensitive mutants (Kuebler and Tanouye, 2002). This reduction could also be achieved by different mutations, which were hence termed seizure suppressors (Saras et al., 2016).
To screen for temperature-related neuronal defects, a reliable way is through a heated water bath, in which flies collected into empty vials can be placed (Burg and Wu, 2012). At a water temperature of ~40°C, seizure-like behavior and paralysis can be observed in susceptible flies as the temperature in the vial increases. Time until seizure or paralysis occurrence, as well as recovery time can be compared. Alterations to seizure susceptibility via genetic or pharmacological manipulation can lead to a more heat-resistant phenotype or a faster recovery. In Drosophila models of GEFS+ and DS, seizure-like behavior and paralysis occurred at elevated temperatures. A faster onset could be observed in the DS model, corresponding to the increased severity of the variant in patients (Sun et al., 2012; Roemmich et al., 2021). Mutations in other genes can induce non-seizure-related paralysis at elevated temperatures. A notable example is the shibire mutant shits, which encodes the Drosophila Dynamin orthologue Shibire. At high temperature, the mutant Shibire protein adopts an inactive conformation. As a consequence, there is a block of synaptic vesicle scission, resulting in paralysis (Kosaka and Ikeda, 1983; Hill et al., 2001). A return to lower temperatures allows the mutant Shibire protein to adopt its normal conformation and restores function. Interestingly, overexpression of shits in neurons of bang-sensitive flies leads to a suppression of mechanically induced seizures at an elevated temperature, as endocytosis becomes impaired (Kroll et al., 2015b).
These assays cover only a portion of behavioral assays in Drosophila. The behavior of flies can also be assessed in terms of activity, locomotion, memory, or social behaviors (Nichols et al., 2012). For instance, sleep disturbances and changes in locomotor activity were observed in a knock-in model of human GEFS+ (Petruccelli et al., 2015; Mituzaite et al., 2021). Also, flies deficient for sif exhibited severe climbing defects besides seizure-susceptibility (Lu et al., 2022). Follow-up investigations of a newfound phenotype with different means are therefore necessary for a comprehensive description.
4.2.2. Investigating neuronal function directlyElectrophysiological recordings allow a more detailed investigation of disturbed neuronal function in Drosophila. Seizure-like activity can be induced in all flies, regardless of genetic background, by direct electrostimulation of the brain (Howlett and Tanouye, 2009). To achieve this, electrodes are inserted in both hemispheres of the brain and a short, high-frequent electric stimulus is applied. Depending on the fly’s susceptibility to seizures, different voltages are required to induce a seizure. In bang-sensitive mutants, low voltages are sufficient to induce seizure-like activity, while wild-type or seizure-resistant mutants require higher voltages for seizure induction (Lee and Wu, 2002). Electrophysiological recordings can be performed at the flight or leg muscle. These muscles are innervated by the giant fiber (GF) pathway, an electrophysiologically well-characterized system (Allen et al., 2006). A seizure disturbs GF function and leads to a characteristic display of muscle potentials. While low-intensity single pulse stimulation at the brain is sufficient to activate the GF pathway and create a signal at the innervated muscle, this transmission is abolished during and shortly after a seizure. With this approach, the seizure phenotype of different genotypes can be quantitatively investigated. This includes the voltage needed to induce seizure-like activity, as well as the recovery time, until GF function is restored. Compared to behavioral assays, this method requires a more complex experimental setup and is less suitable for high-throughput applications. To investigate changes in neuronal function in a broader matter, electroretinograms (ERG) can be recorded from the compound eye of the fly (Wu et al., 2022). Here, light-invoked potentials from photoreceptors and neurons are measured at the surface of the compound eye. Changes between measured responses between different genotypes can reflect defects caused by mutations. Besides these methods, patch-clamp experiments at neurons of the adult fly or larvae are possible as a readout for the activity in individual neurons (Marley and Baines, 2011; Sun et al., 2012).
4.2.3. Optogenetic approachesA non-invasive approach to induce seizures directly at the brain can be taken via optogenetic tools. In optogenetics, neuronal function is altered by introducing channelrhodopsins (ChR), which are light-sensitive ion channels (Zhang et al., 2007). Upon stimulation with light of a specific wavelength, the channels open and allow for the flow of anions or cations. This way, neuronal function can be directly controlled. Using the expression systems described above, ChR expression can be targeted to specific areas of the nervous system. Expressing a red-shifted ChR carrying an excitatory current in the brain of several bang-sensitive mutants allows light-activated seizure induction in these flies (Saras et al., 2017). On the other hand, activation of ChR limited to mushroom body neurons was sufficient to induce seizure-like activity in parabss1 flies. This technique offers a way to pinpoint centers of seizure generation in the fly brain. Optogenetic manipulations can not only be utilized for direct seizure induction in adults but also to alter neuronal function of flies during development. DEEs carry a developmental component, which is closely linked to their etiology (Specchio and Curatolo, 2021). A better understanding of variants in disease-causing genes during development will provide a better understanding of disease etiology and allow for a rational design of therapeutic strategies to treat these syndromes. While embryogenesis is challenging to study in mammals, the development of Drosophila can be easily observed and manipulated. Expression and activation of ChR pan-neuronally during the development of wild-type flies has been shown to permanently alter network activity and to induce persistent seizure-like activity even in adult flies (Giachello and Baines, 2015). Further limiting these network alterations to specific, critical periods in development has also been sufficient to create this phenotype. Longitudinal administration of ASMs alleviated the seizure phenotype. Besides optogenetic approaches, limiting genetic alterations to these critical periods can lead to a seizure-phenotype as well, highlighting the importance of development-specific effects (Horne et al., 2017).
Besides ChR activation by light, gene expression can also be induced by light (Chan et al., 2015; Lee et al., 2017). Here, photosensitive proteins like cryptochrome are linked to effectors, which drive gene expression following a cascade of interactions after light exposure. As a tool combining light-sensitivity with the GAL4/UAS system, PhotoGal4 offers a light-dependent gene expression system which can be used in conjunction with established UAS-lines (de Mena and Rincon-Limas, 2020). The plant-derived phytochrome B is fused to the GAL4-DNA binding domain (GAL4-DBD), while the phytochrome-interacting factor 6 is fused to a VP16 activator domain (VP16-AD). Exposure to red-light allows for a conformational change of phytochrome B and subsequent binding to the phytochrome-interacting factor 6, which brings together the GAL4-DBD and VP16-AD and induces gene expression. Illumination with light in the far-red spectrum reverts phytochrome B to an inactive form, thereby stopping the interaction and gene expression rapidly. A drawback of this method is the requirement of an exogenous chromophore. Another tool that utilizes light-sensitive proteins fused to a GAL4-DBD or AD is ShineGal4 (di Pietro et al., 2021). Here, DBD and AD are brought together by the interaction of “magnet photoswitches”. These proteins act like magnets and heterodimerize upon exposure to blue light (Supplementary Figure 1). By utilizing the GAL4/UAS system, these systems are “backwards compatible”. This expands the possibilities of investigating gene function at precise points in time during development or in the adult fly.
4.2.4. Detection and visualization of neuronal activityTo further understand network functions and alterations, neuronal activity in flies can be visualized using several strategies (DeNardo and Luo, 2017). Calcium imaging has been extensively used as a readout of neuronal activity (Grienberger and Konnerth, 2012). As calcium is an essential cellular messenger, fluctuations can be directly correlated to neuronal activity and visualized through chemical or genetically encoded calcium indicators (GECIs). Since conventional GECIs require monitoring during behavioral tasks, their use in behavioral applications becomes challenging. For example, loss of the gene cpes leads to light-induced seizure behavior, which was also visible as an increase in neuronal activity in a calcium imaging approach in the fly brain (Kunduri et al., 2018). In a simpler setup suitable for larger screens, the calcium-modulated photoactivatable ratiometric integrator (CaMPARI) has been used to label active neurons in freely moving flies (Fosque et al., 2015). CaMPARI is a calcium indicator that uses two components to display neuronal activity. Under conditions where calcium and UV light are available, CaMPARI undergoes a molecular switch and permanently changes from green to red fluorescence. Using UV light only during a defined experimental stimulus, neurons activated in this temporal window can be labeled for functional mapping of neural networks (Zolnik et al., 2017). The technique has been used in Drosophila to mark acid-sensing neurons in flies, while they were allowed to move freely in an environment containing either acid or neutralized acid (Edwards et al., 2020). In the context of seizure-like activity, this method might help to further investigate which neuronal networks are crucial during seizure generation. The role of ASMs and other factors modulating seizure-like activity can also be assessed in a visual manner via calcium imaging. Preparations of the larval nervous system provided another possibility for large-scale screening of ASMs, as they influenced the spatial–temporal patterns of waves of calcium activity between segments of the ventral nerve chord (Streit et al., 2016).
Other approaches to trace calcium in neurons are based on transcriptional activators. In the presence of calcium, they drive expression of downstream targets like GFP, which then accumulates in recently active neurons. In the CaLexA system (calcium-dependent nuclear import of Lex A), the calcium-responsive transcription factor NFAT (nuclear factor of activated T cells) is fused to LexA and VP16 (Masuyama et al., 2012). The expression of the fusion protein is achieved via GAL4/UAS. Presence of calcium leads to dephosphorylation of LexA-VP16-NFAT by the calcium/calmodulin-dependent phosphatase calcineurin and subsequent translocation of the protein to the nucleus, where the LexA domain can drive expression of a reporter. In a similar approach, the TRIC system (transcriptional reporter of intracellular Ca2+) utilizes calcium-based binding of calmodulin (CaM) to its target peptide as a reporter of activity (Gao et al., 2015). CaM is fused to a transcriptional activation domain, while its target peptide is fused to a DNA binding domain of a transcription factor like GAL4. Binding of the fusion proteins then facilitates gene expression. The system acts on a slower timescale, reporting activity changes over long periods of time. Both the CaLexA and the TRIC system allow for calcium-based genetic access to active neurons and therefore modulation of those. Similar to optogenetic tools, where disruptions in neuronal activity have already been shown to alter the seizure phenotype under certain conditions (Giachello and Baines, 2015), these tools could be used to directly alter gene function during development.
Overall, Drosophila provides a versatile toolkit to induce, quantify and visualize seizure-like behavior. In combination with the vast genetic toolkit, large-scale screens to investigate the role of genes in neuronal function become possible.
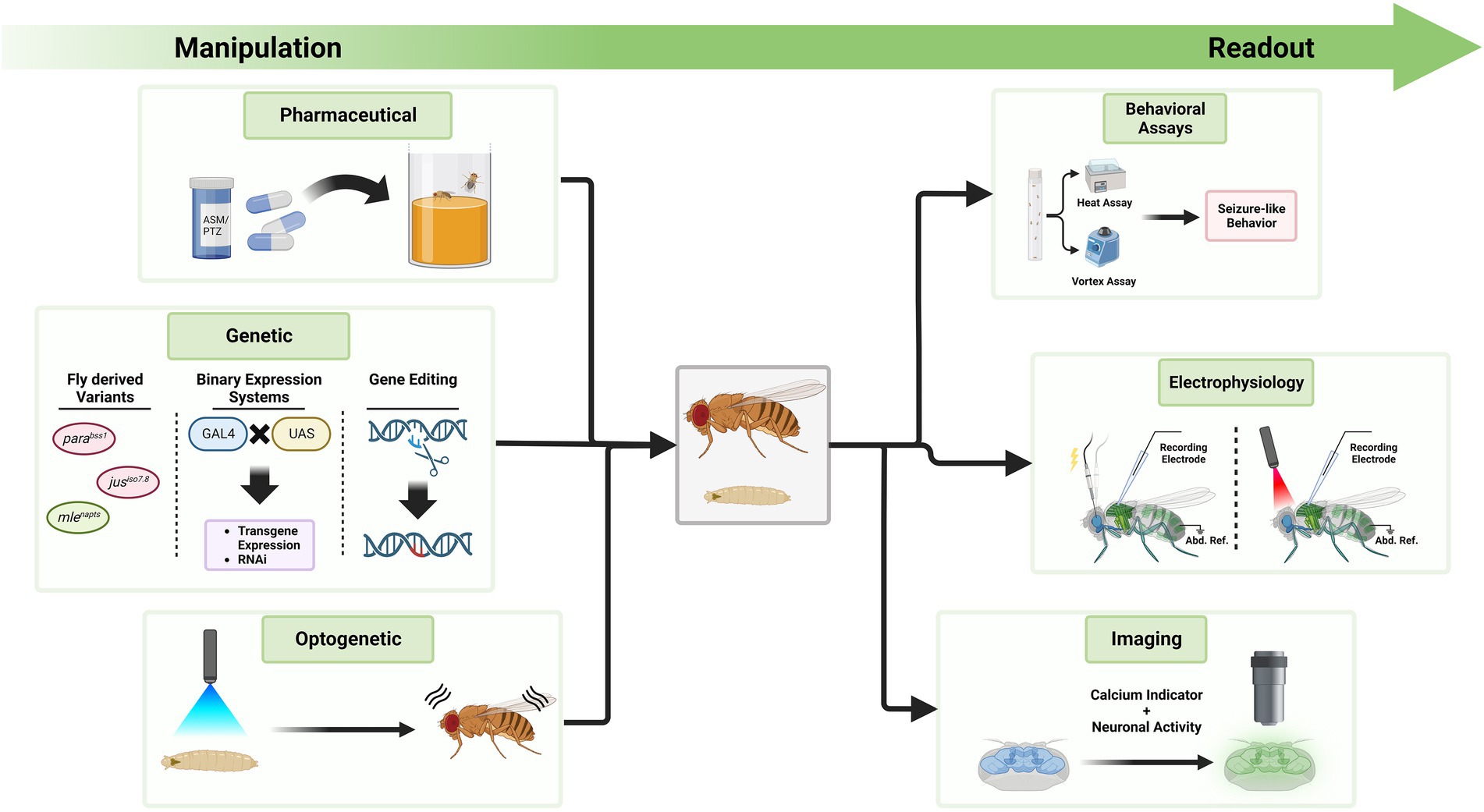
4.3. Developments in Drosophila as a model for epilepsy and future possibilitiesWith the extensive methods of genetic manipulation and readouts available, Drosophila is a suitable model organism for investigations of genetic epilepsies (Figure 4). Here, we outline modern techniques to investigate seizures in Drosophila and their possible applications in the future. Since the number of disease-associated variants of unknown effect size is rising, methods are needed to efficiently study functional consequences of such variants.
Figure 4. Exemplary tools to manipulate seizure-susceptibility in Drosophila and to assess a seizure-like phenotype. The Drosophila toolkit allows to manipulate seizure-susceptibility in flies and to assess their phenotype in relation to seizures. On the left side, different methods are displayed through which Drosophila can be manipulated. Feeding of compounds like ASMs or PTZ, genetic manipulation, or optogenetic manipulation can exacerbate or decrease seizure-susceptibility. Fly-derived variants increasing seizure-susceptibility are color coded in red, those decreasing it in green. To investigate the effect of these alterations, methods on the right side can be used. These include behavioral assays, electrophysiological measurements and imaging approaches. Seizure induction during electrophysiological measurements can be achieved via electrostimulation at the brain or light stimulation if neurons express channelrhodopsins. Activity is measured at the flight muscle with a reference electrode in the abdomen (Abd. Ref.). Techniques on either side can be utilized simultaneously, depending on the research question at hand.
4.3.1. Future potential of large-scale screeningLarge-scale screens can be performed to study the effects of either genetic variance or compound treatment. Such genetic screens played an important role in forward-genetic approaches in the past, leading to the discovery of many seizure-related genes in Drosophila (
留言 (0)