
記住我








Available online 27 January 2023, 106375
 Author links open overlay panelABSTRACT
Author links open overlay panelABSTRACTThe activation and proliferation of hepatic stellate cells (HSCs) are critical processes for the treatment of liver fibrosis. It is necessary to identify effective drugs for the treatment of liver fibrosis and elucidate their mechanisms of action. Metformin can inhibit HSCs; however, no systematic studies demonstrating the effects of metformin on mitochondria in HSCs have been reported. This study demonstrated that metformin induces mitochondrial fission by phosphorylating AMPK/DRP1 (S616) in HSCs to decrease the expression of α-SMA and collagen. Additionally, metformin repressed the total ATP production rate, especially the production rate of ATP produced through mitochondrial oxidative phosphorylation, by inhibiting the enzymatic activity of complex I. Further analysis revealed that metformin strongly constrained the transcription of mitochondrial genes (ND1–ND6 and ND4L) that encode the core subunits of respiratory chain I. Upregulation of the mRNA expression of HK2 and GLUT1 slightly enhanced glycolysis. Additionally, metformin increased mitochondrial DNA (mtDNA) copy number to suppress the proliferation and activation of HSCs, indicating that mtDNA copy number can alter the fate of HSCs. In conclusion, metformin can induce mitochondrial fragmentation and low-level energy metabolism in HSCs, thereby suppressing HSCs activation and proliferation to reverse liver fibrosis.
Section snippetsINTRODUCTIONChronic liver injury leads to liver fibrosis, which is characterized by abnormally activated hepatic stellate cells (HSCs) and excessive extracellular matrix (ECM) accumulation in the liver (Kisseleva and Brenner, 2021). Liver fibrosis can progress to cirrhosis, and hepatocellular carcinoma mostly develops in the fibrotic liver (Myojin et al., 2021). At least 7 million Chinese individuals live with developed liver cirrhosis, and their prognosis and quality of life are very poor (Xiao et al.,
Cell cultureThe human HSC line LX2 and rat HSC line T6 were obtained from Shanghai Anwei Biotechnology Co., Ltd. The cells were cultured according to culture conditions recommended by ATCC (https://www.atcc.org/). The cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM, Life Technologies) supplemented with 10% FBS at 37ºC in a humidified 5% CO2 incubator.
Cell proliferation assayCell counting kit-8 (CCK8, DOJINDO) was used to assess cell proliferation. LX2 and T6 cells were seeded in 96-well plates at a density of 5×103
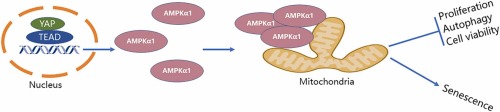
Metformin induced mitochondrial fission in hepatic stellate cellsAMPK is required for mitochondrial fragmentation in response to the constantly changing energy requirements (Roy et al., 2015, Toyama et al., 2016). To verify the role of AMPK in mitochondrial division in HSCs, the AMPKα1 gene was knocked down in LX2 cells. The results revealed that mitochondria did not progress from the elongation to the fragmentation stage in the absence of AMPKα1 (Fig. 1 A, 1B), suggesting that AMPK activity is essential for regulating the morphological characteristics of
DiscussionThis study demonstrated that metformin regulates mitochondrial morphology and alters cellular bioenergetics to decrease HSC activation/proliferation and collagen accumulation. In vitro experiments showed that metformin induced mitochondrial fragmentation by upregulating phosphorylation of Drp1 at the S616 residue and downregulating MFN1/2 and OPA1 simultaneously. Additionally, metformin altered cellular bioenergetics by reducing OXPHOS in HSCs by inhibiting the enzymatic activity of respiratory
ConclusionsMetformin regulates mitochondrial morphology and energy metabolism through multiple mechanisms to reduce HSC activation/proliferation and collagen accumulation. These regulatory effects are mainly achieved by inducing mitochondrial fission and suppressing ATP production and OXPHOS in HSCs. Metformin-induced mitochondrial fragmentation depends on AMPK/DRP1 activation and MFN1/2 suppression. Quantification of mtDNA transcription and enzymatic activity revealed that the inhibition of oxidative
CRediT authorship contribution statementYing Su:Conceptualization, Investigation, Visualization and Writing; Meili Wang: Data Curation and Project administration; Kehan Ren and Danmei Zhou: Writing – Review and Editing; Chenjian Hou: Formal analysis and Validation; Xiaoli Liu, and Shanyu Zhao: Resources; Xiuping Liu: Conceptualization, Supervision and Funding acquisition. All authors read and approved the final manuscript.
Author contributionsYing Su: Conceptualization, Investigation, Data visualization, and Writing; Meili Wang: Data Curation and Project administration. Kehan Ren and Danmei Zhou: Writing – Review and Editing. Chenjian Hou: Formal analysis and Validation. Xiaoli Liu, and Shanyu Zhao: Resources. Xiuping Liu: Conceptualization, Supervision, and Funding acquisition. All authors have read and approved the final manuscript.
Uncited references(Feng et al., 2014, Maria Peiris-Pagès, G.B., Federica Sotgia and Michael P. Lisanti, 2018, Park et al., 2011, Park et al., 2011, Rodrigue Rossignol, R.G., Robert Aggeler, Kunihiro Yamagata, S. James Remington, and Roderick A. Capaldi, 2004, Zhen et al., 2018)
Acknowledgements and fundingThis study was supported by the Shanghai Natural Science Foundation (No.: 21ZR1450300); Medical Specialty funded by Minhang, Shanghai (No.: 2020MWFC02); and Key Medical Specialty funded by Shanghai Fifth People’s Hospital, Fudan University (No.: 2020WYZDZK11).
Conflict of interestThe author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
View full text© 2023 Elsevier Ltd. All rights reserved.
留言 (0)