

記住我
More than 35 years ago, and shortly following the discovery of rat brain 5-HT2 (now considered 5-HT2A) serotonin receptors, phenylisopropylamine analogues were identified as a novel 5-HT2 receptor chemotype (Shannon et al., 1984). In fact, some evidence for this was available from earlier studies using peripheral 5-HT receptor preparations (Glennon et al., 1978) (i.e., rat fundus 5-HT receptors; years later, initially termed 5-HT2F—“F” for rat “fundus”—receptors, these are now considered 5-HT2B serotonin receptors (Kursar et al., 1992)). Certain methoxy-substituted phenylisopropylamines, particularly 2,5-dimethoxy analogs 1 with different R substituents at the aryl 4-position, displayed varying and nanomolar affinities for rat fundus and, later, rat brain 5-HT2 receptors (Seggel et al., 1990), and their affinities were correlated both with their discriminative stimulus properties in rats and their human hallucinogenic potencies (reviewed: Glennon 1996).
In an effort to understand how the 4-position R-substituents of 1 influence 5-HT2 receptor affinity, an extended series of analogs (n = 24) was prepared and a QSAR study conducted (Glennon and Seggel 1989; Seggel et al., 1990). Using rat brain frontal cortex homogenates with the 5-HT2 receptor antagonist [3H]ketanserin as radioligand, QSAR studies employing rat 5-HT2 binding data revealed, for these two dozen analogues of 1 with affinities spanning over a >10,000-fold range (i.e., Ki = 2.5 to 26,000 nM), that affinity was related to the lipophilicity (i.e., π value) and electron withdrawing character (i.e., σp) of the 4-position substituent (Glennon and Seggel 1989). That is, increasing the lipophilicity and the electron withdrawing character of the 4-position R group seemed responsible for rat 5-HT2 receptor affinity. There was also a hint that the “overall size” of the 4-R substituent might play a role in binding (Glennon and Seggel 1989).
Follow-up binding studies were conducted on a sub-set of representative 2,5-DMA (1a) (i.e., 1; R = H for 2,5-DMA) analogues from the rat brain homogenate studies with cloned human 5-HT2A serotonin receptors using [125I]DOI, a 5-HT2 agonist radioligand (i.e., radioiodinated 1 where R = 125I) (Nelson et al., 1999); data for compounds common to the two investigations discussed here are shown in Table 1-A). Human 5-HT2B receptor binding data were also reported in the same investigation. Neither functional nor QSAR studies were performed with the human data in the previous investigation.
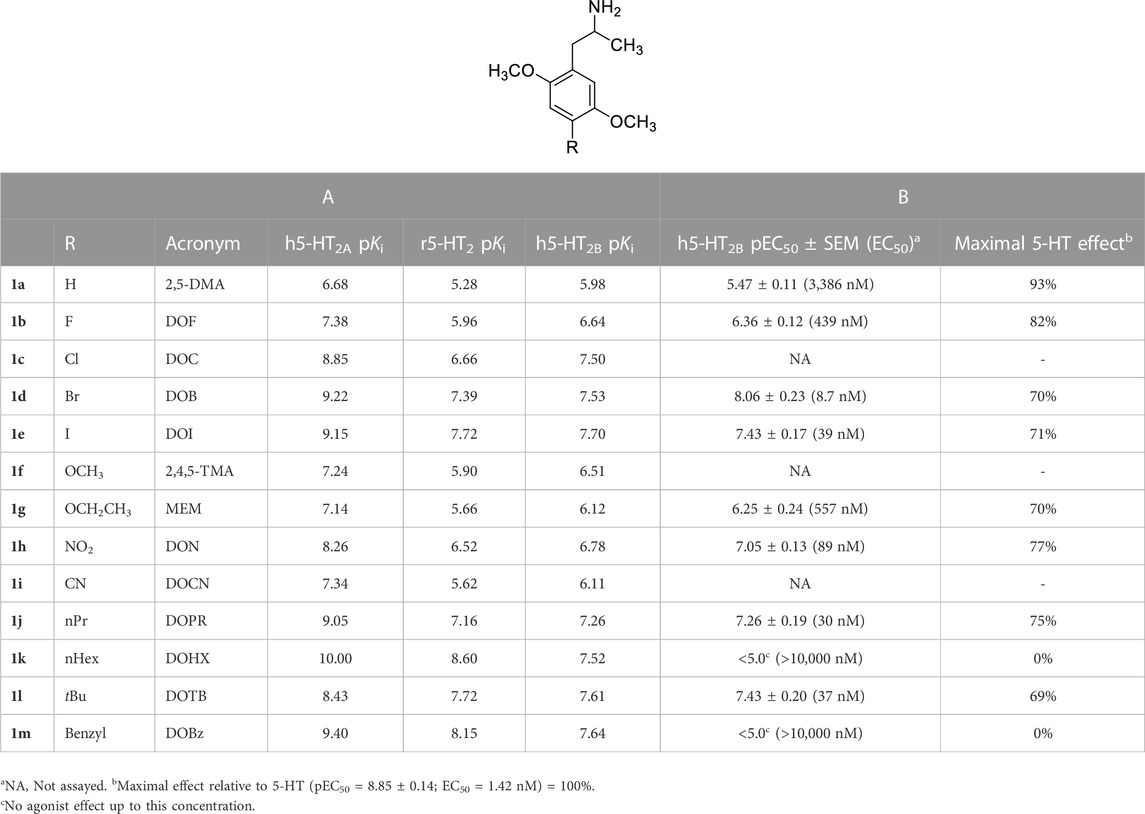
TABLE 1. A: Published radioligand binding data (pKi values) for a series of analogues 1 common to human 5-HT2A vs. [125I]DOI, human 5-HT2B vs. [3H]5-HT (Nelson et al., 1999) and rat 5-HT2 vs. [3H]ketanserin (Seggel et al., 1990) receptor studies, and B: agonist potencies (EC50 values) and maximal 5-HT-related effect (5-HT = 100%) at human 5-HT2B (h5-HT2B) receptors as determined in this study.
The psychoactive properties of classical hallucinogens (including certain phenylisopropylamines 1) appear to involve their agonist action at 5-HT2A serotonin receptors (reviewed: Glennon 1996), whereas activation of 5-HT2B receptors can lead to several serious cardiovascular problems (e.g. vavulopathy) (Rothman et al., 2000; Setola et al., 2005; Roth 2007; Elangbam 2010). The goals of the current study were: i) to determine if the SAR of analogues 1 (Table 1) at human 5-HT2A receptors are related to their SAR at rat 5-HT2 receptors, ii) to affirm (or counter) previous rat 5-HT2 binding QSAR findings for common analogues 1 by examining their human 5-HT2A receptor affinities, iii) to investigate the possibility that human 5-HT2A SAR and QSAR results might inform human 5-HT2B receptor action, and iv) to determine if 5-HT2B receptor affinity and binding QSAR findings are a reliable predictor of 5-HT2B agonist action. These results could have substantial translational or clinical ramifications on the abuse of psychoactive 5-HT2 receptor agonists regarding their potential for producing adverse cardiac events.
2 Materials and methods2.1 MaterialsCompounds examined in the functional assays were available as their hydrochloride salts from earlier synthetic studies conducted in our laboratory.
2.2 5-HT2B receptor functional activity using Ca2+ mobilization assayA stable cell line was generated expressing the human 5-HT2B receptor using the Flp-In T-REx system (Thermo Fisher) (Younkin et al., 2016; Steele et al., 2021). Briefly, 5-HT2B receptor coding plasmid was obtained from cDNA Resource Center (cat # HTR02B0000). The 5-HT2B receptor cDNA was subcloned into the pcDNA/FRT/TO expression plasmid, and was co-transfected with pOG44 plasmid (coding the Flp recombinase) in Flp-In T-REx 293 cells. Cells were selected using 100 μg/mL of hygromycin and resistant cells were expanded and stored in liquid N2 for later use. For experimentation, stable cell lines were plated in Matrigel-coated 96-well imaging plates in Dulbecco’s Modified Eagle Medium (DMEM) containing 10% fetal bovine serum (FBS) and 5% penicillin/streptomycin, and the medium was supplemented with doxycycline (1 μg/mL) to upregulate the expression of the receptor 3 days before the experiment. Then, cells were loaded with Fura 2-AM for 30 min in Ca2+ imaging solution (IS) consistent of 130 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, 10 glucose (in mM, pH adjusted to 7.4). Ca2+ measurements were performed in IS media at room temperature under constant perfusion using the equipment previously described in Ruchala et al. (2021). Fura2 was visualized using epifluorescence microscopy at two excitation wavelengths (340 nm and 380 nm) and a common single emission (510/40 nm). Ratio images were recorded, and the effect of each compound was tested at a single concertation per well and compared to the effect of 1 µM 5-HT. Antagonists were tested by inhibiting 10 nM 5-HT Ca2+ signal, preincubating them for 45 s in combination with 5-HT. Three wells containing cells were analyzed for each concentration per experiment day. Two experiments were conducted for each drug testing at least 6 wells per experimental point in total. Data are depicted as mean ± SD. All data were processed used Fiji software by ImageJ2 which allowed manual selection of cells and measurement of fluorescence. Logarithmic concentration-response curves were generated using GraphPad Prism 8. Curves were generated with a Hill slope of 1.0 to allow easier comparison of potencies of the various compounds analyzed.
2.3 PlottingData were plotted and correlations and QSAR regression analysis and multiple linear regression analysis were conducted using GraphPad Prism 9.03.
3 Results3.1 Relationship between rat and human 5-HT2 receptor bindingFor 13 compounds common to the rat and human 5-HT2A receptor studies (Table 1-A), affinities were higher at the human 5-HT2A than at rat 5-HT2 receptors, but there was a significant correlation between their receptor affinities (r = 0.942; Supplementary Figure S1; SI) despite differences in methods, species, and radioligands employed. Likewise, there was a significant correlation (r = 0.916) between h5-HT2B and rat 5-HT2 receptor affinities (Table 1-A) for these same 13 compounds (Supplementary Figure S2; SI).
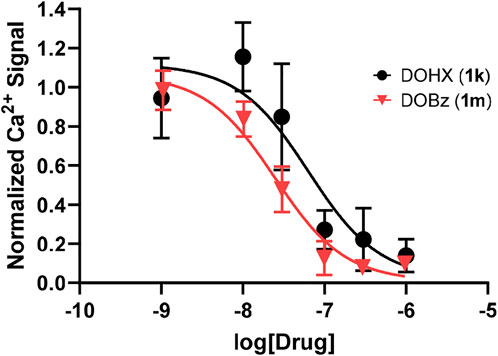
3.2 5-HT2B receptor affinity and functional activityThe 1 analogs bind at 5-HT2B receptors (Table 1-A). Ten of the compounds in Table 1-B (1a, 1b, 1d, 1e, 1g, 1h, 1j, 1k, 1l, 1m) were examined in comparison with 5-HT for their functional activity at human 5-HT2B receptors expressed in HEK 293 (Flp-In T-REx) cells. The compounds were selected on the basis of their range in h5-HT2B receptor affinities, and the diversity of the lipophilic and electronic nature of their 4-position substituent. Initially, the compounds were screened at 10,000 nM; those showing agonist action at a concentration below 10,000 nM were then further evaluated by examining their concentration-response relationships (Table 1-B; Figure 1). They displayed reduced efficacy as agonists (i.e., ca. 0.7–0.9, excluding 1k and 1m; Figure 1) relative to serotonin, and were substantially less potent than 5-HT (EC50 = 1.42 nM) with the most potent analog, DOB (1d), being about 6-fold less potent than 5-HT (Table 1-B). Two of the compounds, 4-hexyl analogue 1k and 4-benzyl analogue 1m, failed to produce an agonist effect at 5-HT2B receptors at the highest concentration examined (10,000 nM), and both behaved as antagonists of the 5-HT-mediated response (Figure 2).
FIGURE 1. Concentration-response curves for eight agonists (Table 1) normalized to 1 µM of the endogenous ligand (5-HT) 5-HT2B response.
FIGURE 2. 5-HT2B receptor antagonist data with 1k (DOHX) pIC50 = 7.2 ± 0.18 (63.6 nM) and 1m (DOBz) pIC50 = 7.61 ± 0.08 (24.6 nM) in the presence of 10 nM of 5-HT.
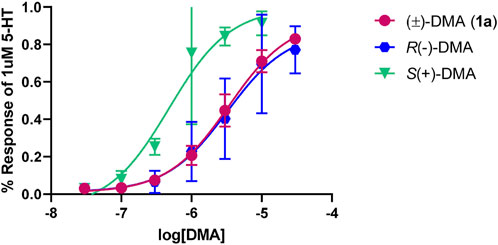
The individual optical isomers of the parent member of the series, 2,5-DMA (1a), were examined at the 5-HT2B receptor; it was found that S (+)1a was about six times more potent (pEC50 = 6.32 ± 0.15, EC50 = 480 nM; 100% 5-HT effect) than its R (−) enantiomer (pEC50 = 5.49 ± 0.16, EC50 = 3,236 nM; 87% 5-HT effect) (Figure 3).
FIGURE 3. Concentration-response curves for 2,5-DMA (1a) and its optical isomers at the 5-HT2B receptor, normalized to 1 µM of the endogenous ligand (5-HT) response.
3.3 QSAR studiesUsing the previously published h5-HT2A receptor binding data from Table 1-A, affinity was found to be correlated with lipophilicity of the 4-position substituent (i.e., π; r = 0.798) alone, and with lipophilicity plus σp (r = 0.917). Whole molecule Volume (Å3), as calculated using SybylX2.1.1 (see Table S1 in SI), was also examined for the compounds in Table 1-A, but volume was found to be inter-correlated with the substituent constant π (r = 0.857). For comparison, rat 5-HT2 binding data for this same series of 13 compounds (Table 1-A) also was found to correlate with (π; r = 0.897) alone, and with lipophilicity plus σp (r = 0.951). A similar QSAR analysis was performed with the h5-HT2B binding data shown in Table 1-A. Affinity was found to be correlated with lipophilicity (i.e., π; r = 0.750) alone, and with lipophilicity plus σp (r = 0.812). However, the contribution of the σp term was not statistically significant.
4 DiscussionConsiderable rat brain homogenate 5-HT2 receptor binding data for various phenylisopropylamines 1 and other serotonergic compounds such as, for example, phenylethylamines, tryptamines, β-carbolines, and ketanserin analogs have been published. Once human 5-HT2A receptors were identified and cloned, it was necessary to determine if binding data for agents at the former rat population reflected data from the latter human studies. Indeed, some studies have already used rat 5-HT2 binding data for phenylisopropylamines as a surrogate for 5-HT2A receptor 3D-QSAR studies (e.g. Schulze-Alexandru et al., 1999). Despite differences in species (rat versus human), radioligands (antagonist [3H]ketanserin versus agonist [125I]DOI), and potential methodological differences, there was no qualitative difference in receptor affinity of the agents examined here. True, this information might not be extrapolated at the current time to non-phenylisopropylamines. However, as might have been expected, on the basis of the use of an agonist radioligand, human 5-HT2A receptor affinities for the examined agents (Table 1-A) were somewhat higher than those for rat 5-HT2 receptors. In addition to the antagonist versus agonist nature of the radioligands employed, differences in affinity might also reflect species (>90% sequence homology between rat and human). But, here, this does not seem to make a difference for the phenylisopropylamines (i.e., 1 analogs) examined due the affinity correlation shown in Figure S1 (SI). Encouraging also was that QSAR studies, those published earlier with rat 5-HT2 binding data (Glennon and Seggel 1989) as well as that presented here with human 5-HT2A receptors, both implicated the lipophilicity and, possibly, the electronic nature of the 4-position substituents as being contributors to receptor affinity; that is, increased affinity is associated with increased lipophilicity and, perhaps, electron withdrawing character.
The lipophilic and electronic character of the 4-position substituents of 1 were also implicated in their binding at h5-HT2B receptors. Furthermore, eight of the 10 compounds in Table 1-A, the exceptions being 1k and 1m, behaved as 5-HT2B receptor agonists. Porter et al. (1999) have previously examined the functional action of DOB (1d) and DOI (1e) (i.e., measurement of intracellular calcium levels in CHO cells expressing human 5-HT2B receptors) and found them to be of similar agonist potency and produced 69% and 65% of the effect of 5-HT, respectively. Also, Huang et al. (2009) have demonstrated that DOI (1e) is a potent 5-HT2B receptor agonist in multiple functional assays of receptor activation. However, given the results in Table 1-A and Figure 1, it is evident that functional potency does not appear to be related to 5-HT2B receptor affinity (e.g. compare DOB, 1d, with its hexyl counterpart 1k which have nearly identical affinities) and, hence, because affinity is related to the lipophilic and, less significantly, electronic nature of the substituents, the latter measures cannot be taken as reliable predictors of functional potency. Benzyl analog 1m also failed to produce an effect. A feature that appears to differentiate the demonstrated agonists from the inactive compounds seems to be the “size” of the 4-position substituent; that is, compounds 1k and 1m possess the most lipophilic, and largest (volume-wise), of the substituents examined (see Supplementary Table S1; SI). This will require further investigation.
Although lacking agonist action, the hexyl and benzyl analogues (1k and 1m, respectively) bind at 5-HT2B receptors with high affinity; hence, the possibility exists that they might represent antagonists. This was shown to be the case in Figure 2. These same two compounds had also been shown earlier to lack rat 5-HT2A agonist action and behave as antagonists (Seggel et al., 1990; Glennon 2017).
Very few QSAR studies have been conducted on the binding of phenylisopropylamine derivatives at human 5-HT2B receptors. Nistala (2018) examined 22 5-HT2B receptor ligands, that included the compounds in Table 1, using comparative molecular field analysis (CoMFA) and concluded that the lipophilicity of 4-position substituents make a positive contribution to binding, but that substituents on the terminal amine were detrimental. Hajjo et al. (2010) using a very large and diverse data set (and although some phenylisopropylamines were included, analogs of 1 were not) developed useful in silico QSAR models for the identification of compounds that might bind at 5-HT2B receptors; useful as they might be, the models were “[unable to] distinguish agonists from antagonists” (Hajjo et al., 2010). The present study found that QSAR studies can aid our understanding of h5-HT2B (and h5-HT2A) receptor binding, but are as yet unable to predict 5-HT2B (or 5-HT2A) agonist action.
Overall, the present investigation i) demonstrated that the human 5-HT2A receptor affinities for 13 analogs of 1 parallel their rat 5-HT2 receptor affinities and that, as a consequence, their interactions might share a common SAR, ii) showed that the human 5-HT2B receptor affinities for these same agents parallel their rat 5-HT2 receptor affinities, iii) confirmed that rat 5-HT2 receptor affinity is associated with the increased lipophilicity and, perhaps, electron withdrawing nature of the 4-position substituents of 1, and shows that this relationship also applies to their interactions at human 5-HT2A and 5-HT2B receptors, iv) examined the functional activity of 10 analogs of 1 at human 5-HT2B receptors, and found that 5-HT2B agonist action could not be predicted simply on the basis of receptor affinity or the QSAR studies conducted (i.e., two high-affinity 5-HT2B ligands lacked agonist action in functional assays). On this basis, rat and h5-HT2A receptor binding data might be employed to estimate the affinity, but not the functional activity, of phenylisopropylamines for h5-HT2B receptors. We have previously demonstrated that agonist and antagonist phenylisopropylamines need not share a common SAR and might bind differently at 5-HT2A receptors (Rangisetty et al., 2001); additional studies should now be conducted with phenylisopropylamines that bind at 5-HT2B receptors.
Studies with the phenylisopropylamines fenfluramine and dexfenfluramine (two of the most widely investigated 5-HT2B-associated valvulopathogens) have demonstrated that their duration of use was strongly predictive of adverse cardiovascular events (Hopkins et al., 2003; Dahl et al., 2008). Nevertheless, given that certain analogs of 1 have been found on the clandestine market, and shown here to behave as 5-HT2B receptor agonists, it would seem that persistent or chronic use of such agents might lead to cardiovascular complications. This requires further investigation. Recent studies have shown that certain other phenylalkylamines (Kolaczynska et al., 2019; Luethi et al., 2019) including mescaline analogs (Kolaczynska et al., 2021) can activate 5-HT2B receptors. Additional studies with these (and related) agents to identify binding and functional pharmacophores for 5-HT2B action would appear warranted.
Data availability statementThe original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Author contributionsAll authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
FundingThis work was in partial fulfillment of the doctoral requirements for PH who was supported by the VCU’s CTSA (UL1TR000058) from the National Center for Advancing Translational Sciences) and the CCTR Endowment Fund of Virginia Commonwealth University (RAG, MD).
Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary materialThe Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2023.1101290/full#supplementary-material
ReferencesDahl, C. F., Allen, M. R., Urie, P. M., and Hopkins, P. N. (2008). Valvular regurgitation and surgery associated with fenfluramine use: An analysis of 5743 individuals. BMC Medicine. 6, 34. doi:10.1186/1741-7015-6-34
PubMed Abstract | CrossRef Full Text | Google Scholar
Glennon, R. A. (1996). “Classical hallucinogens,” in Handbook of experimental Pharmacology: Pharmacological Aspects of drug dependence; schuster. Editors C. R. Schuster, and M. J. Kuhar (Basel, Switzerland: Springer-Verlag), 343–371.
CrossRef Full Text | Google Scholar
Glennon, R. A., Liebowitz, S. M., and Mack, E. C. (1978). Serotonin receptor binding affinities of several hallucinogenic phenalkylamine and N,N-dimethyltryptamine analogs. Journal of Medicinal Chemistry. 21, 822–825. PMID: 278843. doi:10.1021/jm00206a022
PubMed Abstract | CrossRef Full Text | Google Scholar
Glennon, R. A., and Seggel, M. R. (1989). “Interaction of phenylisopropylamines with 5-HT2 receptors: A QSAR analysis,” in Bioactive mechanisms. Editor P. S. Magee (Washington, United States: American Chemical Society), 264–280.
CrossRef Full Text | Google Scholar
Glennon, R. A. (2017). The 2014 philip S. Portoghese medicinal Chemistry lectureship: The "phenylalkylaminome" with a focus on selected drugs of abuse. Journal of Medicinal Chemistry. 60, 2605–2628. PMID: 28244748. doi:10.1021/acs.jmedchem.7b00085
PubMed Abstract | CrossRef Full Text | Google Scholar
Hajjo, R., Grulke, C. M., Golbraikh, A., Setola, V., Huang, X-P., Roth, B. L., et al. (2010). Development, validation, and use of quantitative structure-activity relationship models of 5-hydroxytryptamine (2B) receptor ligands to identify novel receptor binders and putative valvulopathic compounds among common drugs. Journal of Medicinal Chemistry. 53, 7573–7586. PMID: 20958049. doi:10.1021/jm100600y
PubMed Abstract | CrossRef Full Text | Google Scholar
Hopkins, P. N., and Polukoff, G. I. (2003). Risk of valvular heart disease associated with use of fenfluramine. BMC Cardiovascular Disorders. 3, 5. PMID: 12801402. doi:10.1186/1471-2261-3-5
PubMed Abstract | CrossRef Full Text | Google Scholar
Huang, X. P., Setola, V., Yadav, P. N., Allen, J. A., Rogan, S. C., Hanson, B. J., et al. (2009). Parallel functional activity profiling reveals valvulopathogens are potent 5-hydroxytryptamine(2B) receptor agonists: Implications for drug safety assessment. Molecular Pharmacology. 76, 710–722. PMID: 19570945. doi:10.1124/mol.109.058057
PubMed Abstract | CrossRef Full Text | Google Scholar
Kolaczynska, K. E., Luethi, D., Trachsel, D., Hoener, M. C., and Liechti, M. E. (2021). Receptor interaction profiles of 4-alkoxy-3,5-dimethoxy-phenethylamines (mescaline derivatives) and related amphetamines. Frontiers in Pharmacology. 12, 794254. PMID: 35222010; PMCID: PMC8865417. doi:10.3389/fphar.2021.794254
PubMed Abstract | CrossRef Full Text | Google Scholar
Kolaczynska, K. E., Luethi, D., Trachsel, D., Hoener, M. C., and Liechti, M. E. (2019). Receptor interaction profiles of 4-alkoxy-substituted 2,5-dimethoxyphenethylamines and related amphetamines. Frontiers in Pharmacology. 10, 1423. PMID: 31849671; PMCID: PMC6893898. doi:10.3389/fphar.2019.01423
PubMed Abstract | CrossRef Full Text | Google Scholar
Kursar, J. D., Nelson, D. L., Wainscott, D. B., Cohen, M. L., and Baez, M. (1992). Molecular cloning, functional expression, and pharmacological characterization of a novel serotonin receptor (5-hydroxytryptamine2F) from rat stomach fundus. Molecular Pharmacology. 42, 549–557. PMID: 1331748.
PubMed Abstract | Google Scholar
Luethi, D., Widmer, R., Trachsel, D., Hoener, M. C., and Liechti, M. E. (2019). Monoamine receptor interaction profiles of 4-aryl-substituted 2,5-dimethoxyphenethylamines (2C-BI derivatives). European Journal of Pharmacology. 855, 103–111. Epub 2019 May 4. PMID: 31063768. doi:10.1016/j.ejphar.2019.05.014
PubMed Abstract | CrossRef Full Text | Google Scholar
Nelson, D. L., Lucaites, V. L., Wainscott, D. B., and Glennon, R. A. (1999). Comparisons of hallucinogenic phenylisopropylamine binding affinities at cloned human 5-HT2A, 5-HT2B and 5-HT2C receptors. Naunyn-Schmiedeberg's Archives of Pharmacology. 359, 1–6. PMID: 9933142. doi:10.1007/pl00005315
PubMed Abstract | CrossRef Full Text | Google Scholar
Nistala, P. (2018). “5-HT2B receptor-mediated cardiac valvulopathy,” (Richmond, United States: Virginia Commonwealth University). MS thesis.
Porter, R. H., Benwell, K. R., Lamb, H., Malcolm, C. S., Allen, N. H., Revell, D. F., et al. (1999). Functional characterization of agonists at recombinant human 5-HT2A, 5-HT2B and 5-HT2C receptors in CHO-K1 cells. British Journal of Pharmacology. 128, 13–20. PMID: 10498829. doi:10.1038/sj.bjp.0702751
PubMed Abstract | CrossRef Full Text | Google Scholar
Rangisetty, J. B., Dukat, M., Dowd, C. S., Herrick-Davis, K., DuPre, A., Gadepalli, S., et al. (2001). 1-[2-Methoxy-5-(3-phenylpropyl)]-2-aminopropane unexpectedly shows 5-HT2A serotonin receptor affinity and antagonist character. Journal of Medicinal Chemistry. 44, 3283–3291. PMID: 11563927. doi:10.1021/jm0100739
PubMed Abstract | CrossRef Full Text | Google Scholar
Rothman, R. B., Baumann, M. H., Savage, J. E., Rauser, L., McBride, A., Hufeisen, S. J., et al. (2000). Evidence for possible involvement of 5-HT2B receptors in the cardiac valvulopathy associated with fenfluramine and other serotonergic medications. Circulation 102, 2836–2841. PMID: 11104741. doi:10.1161/01.cir.102.23.2836
PubMed Abstract | CrossRef Full Text | Google Scholar
Ruchala, I., Battisti, U. M., Nguyen, V. T., Chen, R. Y., Glennon, R. A., and Eltit, J. M. (2021). Functional characterization of N-octyl 4-methylamphetamine variants and related bivalent compounds at the dopamine and serotonin transporters using Ca2+ channels as sensors. Toxicology and Applied Pharmacology. 419, 115513. PMID: 33785354. doi:10.1016/j.taap.2021.115513
PubMed Abstract | CrossRef Full Text | Google Scholar
Schulze-Alexandru, M., Kovar, K-A., and Vedani, A. (1999). Quasi-atomistic receptor surrogates for the 5-ht2a receptor: A 3D-QSAR study on hallucinogenic substances. Quantitative Structure Activity Relationships. 18, 548–560. doi:10.1002/(sici)1521-3838(199912)18:6<548:aid-qsar548>3.0.co;2-b
CrossRef Full Text | Google Scholar
Seggel, M. R., Yousif, M. Y., Lyon, R. A., Titeler, M., Roth, B. L., Suba, E. A., et al. (1990). A structure-affinity study of the binding of 4-substituted analogues of 1-(2,5-dimethoxyphenyl)-2-aminopropane at 5-HT2 serotonin receptors. Journal of Medicinal Chemistry. 33, 1032–1036. PMID: 2308135. doi:10.1021/jm00165a023
PubMed Abstract | CrossRef Full Text | Google Scholar
Setola, V., Dukat, M., Glennon, R. A., and Roth, B. L. (2005). Molecular determinants for the interaction of the valvulopathic anorexigen norfenfluramine with the 5-HT2B receptor. Molecular Pharmacology. 68, 20–33. PMID: 15831837. doi:10.1124/mol.104.009266
PubMed Abstract | CrossRef Full Text | Google Scholar
Shannon, M., Battaglia, G., Glennon, R. A., and Titeler, M. (1984). 5-HT1 and 5-HT2 binding properties of derivatives of the hallucinogen 1-(2,5-dimethoxyphenyl)-2-aminopropane (2,5-DMA). European Journal of Pharmacology. 102, 23–29. PMID: 6479216. doi:10.1016/0014-2999(84)90333-9
PubMed Abstract | CrossRef Full Text | Google Scholar
Steele, T. W. E., Spires, Z., Jones, C. B., Glennon, R. A., Dukat, M., and Eltit, J. M. (2021). Non-conserved residues dictate dopamine transporter selectivity for the potent synthetic cathinone and psychostimulant MDPV. Neuropharmacology 200, 108820. PMID: 34619165. doi:10.1016/j.neuropharm.2021.108820
PubMed Abstract | CrossRef Full Text | Google Scholar
Younkin, J., Gaitonde, S. A., Ellaithy, A., Vekariya, R., Baki, L., Moreno, J. L., et al. (2016). Reformulating a pharmacophore for 5-ht2a serotonin receptor antagonists. ACS Chemical Neuroscience. 7, 1292–1299. PMID: 27385190. doi:10.1021/acschemneuro.6b00162
留言 (0)