
記住我
Potassium (K+) is the major electrolyte component in intracellular fluid (ICF) and is crucial for vital biological functions. Almost all of the total body K+ is located in the ICF, where it plays a crucial role in protein synthesis and cell growth (1, 2). A positive K+ balance is essential for somatic growth in growing children, especially in young infants. However, severe hyperkalemia is life-threatening and it can cause cardiac arrhythmias as well as cardiac arrest. Multiple etiologies can cause hyperkalemia including increased K+ load, renal dysfunction and certain drugs, but endocrine disorders should also be considered. Aldosterone (Ald) is the major mineralocorticoid hormone and it is a vital factor for maintaining electrolyte and water homeostasis in humans. It is produced exclusively in the adrenal zona glomerulosa and plays an important role in regulating the re-absorption of sodium (Na+) as well as the excretion of K+via the distal nephrons (3). Ald secretion is mainly regulated by the renin–angiotensin system.
Ald defects, include impairments in Ald biosynthesis (primary hypoaldosteronism) and aldosterone resistance (pseudohypoaldosteronism). Defects in biosynthesis of Ald are usually related to genetic mutations which can result in a disability to produce enzymes or it can be caused by destruction or hypoplasia of adrenal cortex (4). Primary adrenal insufficiency (PAI) including congenital adrenal hyperplasia (CAH), X-linked adrenal hypoplasia congenita (X-AHC) and Xp21 contiguous gene deletion syndrome, are conditions characterized by a failure to produce cortisol and it is the most common etiology of Ald synthesis defects. Other etiologies such as isolated hypoaldosteronism (IHA) are rarely seen (5, 6). IHA, known as Ald synthase deficiency, is an extremely rare autosomal recessive disorder mainly caused by inactivating mutations of the CYP11B2 gene (7, 8). Inactivating mutations in the CYP11B2 gene can lead to abnormalities of aldosterone synthase which is the enzyme responsible for catalyzing the terminal steps in Ald biosynthesis and converts 11-deoxycorticosterone to Ald.
Ald resistance (9), is characterized by pseudohypoaldosteronism and result in insufficient K+ and hydrogen secretion (10, 11). Pseudohypoaldosteronism (PHA) has been described as three different forms: PHA types 1, 2 and 3 (PHA1, PHA2 and PHA3, respectively). PHA1 and PHA2 are referred to as primary pseudohypoaldosteronism. PHA1 is characterized by either abnormalities of the mineralocorticoid receptor or dysfunction in epithelial Na+ channel which responds to Ald in distal nephrons. Mutations in the mineralocorticoid receptor coding gene, NR3C2, and the epithelial Na+ channel coding gene of subunit genes (SCNN1A, SCNN1B and SCNN1G) are responsible for PHA1 (11).
PHA2 is known as familial hyperkalemic hypertension or Gordon's Syndrome. Mutations in the genes involved in either the pathway of the thiazide sensitive Na + chloride cotransporter (NCC) or renal outer medullary K+ channel (ROMK) are responsible for this disorder. The genes WNK1, WNK4, KLHL3 and CUL3 were shown to cause PHA2 (12–14). PHA3 (which is a secondary PHA or transient PHA), is caused by various pathologies of either the urinary and intestinal tracts or the sweat glands. Urinary tract infections and urinary malformations are the most common cause of PHA3 (15).
Patients with Ald defects typically present with growth retardation, salt-lost, hyperkalemia, hypovolemia and metabolic acidosis. However, rather than episodes of hypovolemia and salt-lost, patients with PHA2 usually present with low-renin hypertension, hyperkalemia and metabolic acidosis (12).
Numerous conditions such as CAH, IHA and PHA may present with similar clinical symptoms. This may result in difficulties producing a specific diagnosis for these conditions. The correct diagnosis is the most essential premise in order to achieve a successful treatment regimen. In this study, we present the clinical and genetic features of children with Ald signaling defects who are associated with hyperkalemia.
Methods Patients and data collectionA retrospective study was conducted at the Pediatric unit of the First Affiliated Hospital of Guangxi Medical University from 1st January 2012 to 1st October 2022. The selection criteria were as follows: (1) clinical signs and symptoms associated with Ald defects, such as recurrent vomiting, growth retardation, hypertension in infants and young children (age under 3 years old). (2) hyperkalemia (serum K+ > 5.0 mmol/L) with or without hyponatremia (serum Na+ < 135.0 mmol/L) and metabolic acidosis. (3) If necessary, a positive genetic test relating to one of the etiologies of Ald defect by using an appropriate molecular genetic testing technology. Diagnosis of a disorder was based on its practical guidelines and for rare cases with no guidelines. This mainly depended on the clinical presentation, and results of biochemical examinations and molecular genetic testing. Patients with hyperkalemia but not due to aldosterone defects such as chronic kidney disease, hyperkalemia secondary to hemolysis, type IV renal tubular acidosis, medication related hyperkalemia were excluded from this study. Meanwhile, the patients with incomplete data were excluded from this study. The following data were collected from patients' records: demography, family history, clinical, biochemical, imaging tests and hormonal features at initial presentation. The results of genetic mutation analysis and treatments were also included.
ResultsForty-seven patients with hyperkalemia were enrolled into this study. Of these, 38 patients were associated with Ald biosynthesis defects and 9 patients had related to Ald resistance.
Patients with primary hypoaldosteronismThirty-eight patients were associated with defects in Ald biosynthesis and 32 of these contributed to CAH due to 21-hydroxylase deficiency. Four patients were clinically diagnosed with primary adrenal insufficiency with either a negative gene analysis or failure to conduct genetic testing. One patient was diagnosed with Xp21 contiguous gene deletion syndrome because of a rearrangement on Xp21 which contains the NROB1 gene (Supplementary Table S1). All of them had hyperkalemia as well as hyponatremia.
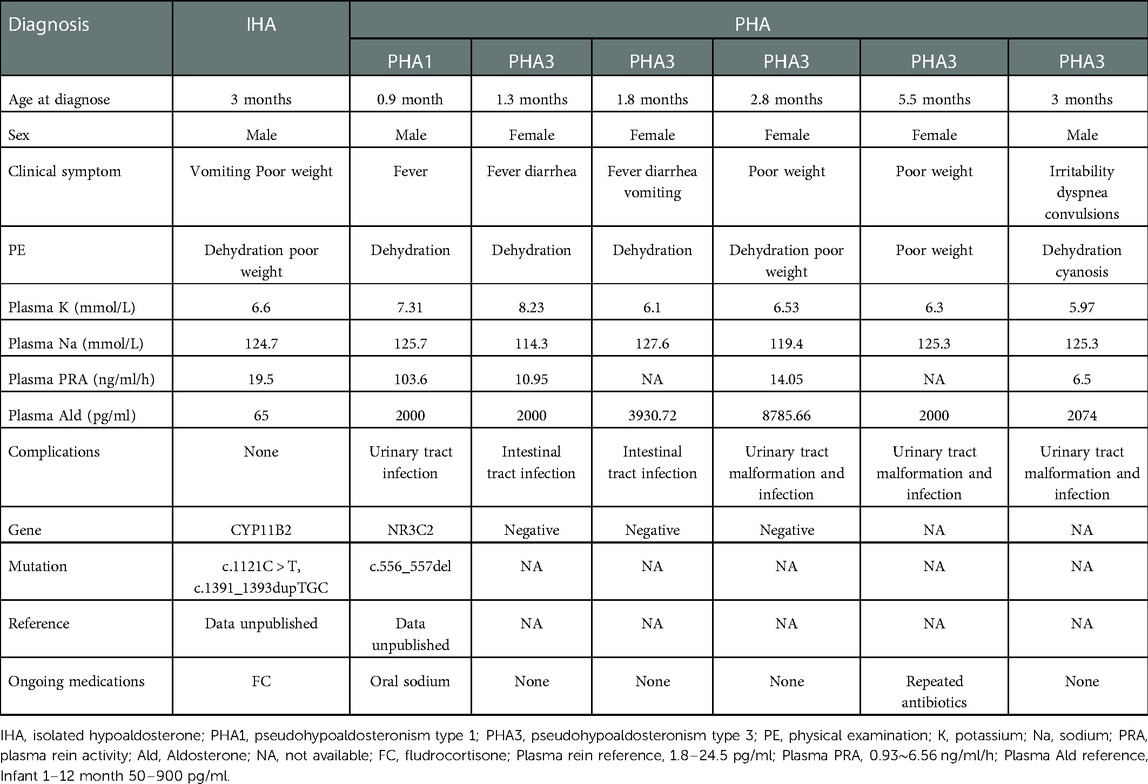
An elevated level of serum adrenocorticotropic hormone (ACTH) combined with a low level of serum cortisol were observed in patients with primary adrenal insufficiency or Xp21 contiguous gene deletion syndrome. Test for hormone showed an elevated level of 17-hydroxyprogesterone (17-OHP) in all patients with CAH. Twenty-two patients with CAH were subjected to genetic analysis and all of them were shown to have mutations in the CYP21A2 gene. In addition, one male infant was diagnosed with isolated hypoaldosteronism (IHA) due to a CYP11B2 gene mutation and his detailed clinical features are shown in Table 1.
Table 1. Characteristics of young children with isolated hypoaldosterone and pseudohypoaldosteronism types 1 and 3.
Patients with pseudohypoaldosteronismAll three types of PHA were all identified in our study, and one boy had PHA1 as well as a urinary tract infection. Five infants were associated with PHA3 which was secondary to the pathologies of either the urinary or intestinal tracts. Patients with either PHA1 or PHA3 all presented with similar clinical symptoms such as poor weight gain, vomiting and fever during infancy and their characteristics are shown in Table 1.
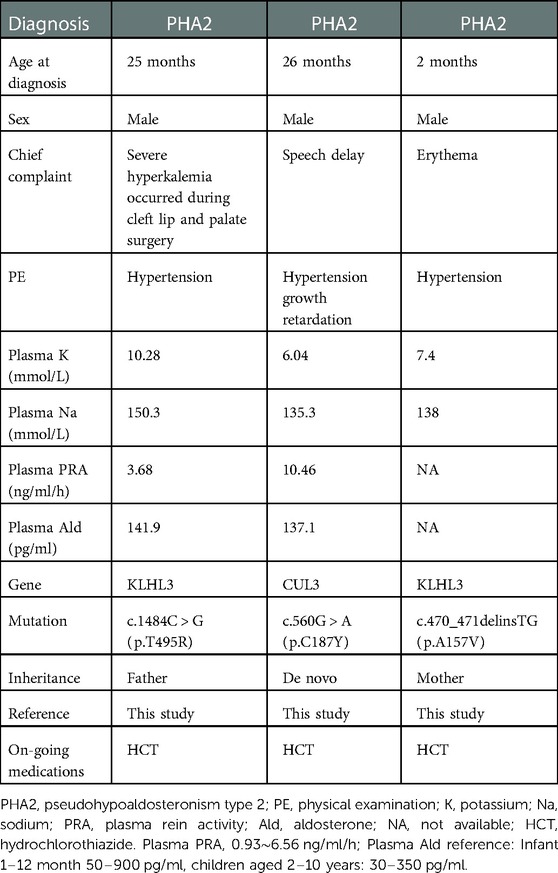
One infant and two young children were diagnosed with PHA2. They came to our hospital with different chief complaints and were incidentally discovered to have hyperkalemia and hypertension and they were finally diagnosed with PHA2. The first child was found to have a novel heterozygous mutation of the KLHL3 gene, c.1484C > G (p.T495R), which was inherited from his father. The second child was observed to have a de novo mutation of the CUL3 gene, c.560G > A (p.C187Y). The third infant was identified to have a novel pathogenic heterozygous mutation of the KLHL3 gene, c.470_471delinsTG (p.A157V), which was inherited maternally. These variants had not been reported by either the ExAC or the Human Gene Mutation Database. Their clinical and genetic characteristics are shown in Table 2. Their family trees and Sanger sequencing are shown in Supplementary Figures S1–S3, respectively.
Table 2. Characteristics of patients with pseudohypoaldosteronism type 2.
DiscussionAld, a potent mineralocorticoid hormone, is synthesized exclusively by the adrenal zona glomerulosa. It can actively enhance Na+ reabsorption and secrete K+ and hydrogen ions via the distal renal nephrons. Ald biosynthesis defects or Ald resistance can result in electrolyte imbalance (hyponatremia with or without hyperkalemia) and metabolic acidosis. In most cases, hyponatremia can be easily identified, because it usually accompanied by some symptoms, such as recurrent vomiting, failure to thrive, dehydration and seizures. However, patients with hyperkalemia may be particularly difficult to recognize as they are often asymptomatic for most of the time. This is especially the case in young children. Severe hyperkalemia can be dangerous, if not identified and treated immediately. It may cause cardiac arrhythmias and arrest and can even lead to death. Hyperkalemia is a rare but severe condition in young children and in most cases, it is seen after difficulties in obtaining adequate blood samples due to hemolysis of these when being taken. However, patients with Ald signaling defects can also present with hyperkalemia and this may ignore by clinicians, especially when it occurs in young children.
Primary adrenal insufficiency, specifically to CAH with concomitant hypoaldosteronism is relatively common in young children. As in our study, this condition accounted for 68.1% (n = 32) of total cases with hyperkalemia (16). All of whom had hyperkalemia as well as hyponatremia during infancy. Steroid tests and genetic analysis can be useful to make a quick and correct diagnosis which can help when conducting genetic counseling to the prenatal mother. In addition to the most common cortisol deficiency associated with Ald defects (CAH due to 21-hydroxylase deficiency), several different etiologies related aldosterone signal defects were identified in this study including Xp21 contiguous gene deletion syndrome, IHA and PHA.
IHA is characterized by defects in Ald biosynthesis due to the Ald synthase deficiency. Most patients usually show a low plasma level of Ald. However, a normal or even a high level of plasma Ald was observed in some patients with IHA (17, 18). PHA1 is characterized by mineralocorticoid resistance due either to an inactive mutation in the mineralocorticoid receptor or the epithelial Na+ channel. Patients with PHA1 usually presented with a high level of serum Ald due to mineralocorticoid resistance. A marked elevated plasma Ald level may be a useful biomarker to distinguish the PHA1 from IHA. However, not all patients with PHA1 show elevated aldosterone levels (9). Actually, it is difficult to explain the circulating hormone levels observed during young childhood of these patients, especially in early infancy. If necessary, either repeated hormone testing or conducting serum and urine steroid precursors tests as well as performing a genetic analysis should be considered.
PHA2 is a rare hereditary disease and is associated with an over-activating thiazide-sensitive NaCl co-transporter in the distal nephrons, resulting in excessive Na+ resorption, which decreases the excretion of K+ and hydrogen (13, 19). It is characterized by hypertension, hyperkalemia, hyperchloremic metabolic acidosis and normal renal function. Plasma renin levels are usually suppressed and aldosterone levels can be variable but are relatively low due to the hyperkalemia (20). Blood pressure, which may be ignored in children, especially in early infancy, can be an important hint for establishing the diagnosis of PHA2. In our study, we carefully checked the blood pressure in all the young children and infants who presented with hyperkalemia without hyponatremia and finally we were able to make a correct diagnosis.
Ald is essential for K+ excretion and Na+ retention in the kidneys, salivary glands, sweat glands and colon. PHA3, known as reversible secondary pseudohypoaldosteronism, is described mainly because it is caused by urinary tract infections and anomalies during infancy. Watanabe et al. (21) reviewed 60 cases of secondary PHA and found all of them occurred at less than 7 months of age. In addition, all the patients suffered from urinary tract infections with or without urinary tract malformations. Patients with PHA3 due to pathology of the intestinal tract are relatively rare. Several patients who underwent operations for colectomy or small bowel resection or jejunostomy or ileostomy in adults and infants have been reported (22–24). However, patients with gastrointestinal losses-related PHA3 due to intestinal infections had been rarely reported. In our study, two infants who had not undergone any gastro-enteric operations were found to have PHA3 due to severe diarrhea associated with intestinal infections. Remarkably, it was difficult to distinguish PHA3 from PHA1 only by clinical symptoms and biochemical testing. This was particularly the case for patients with PHA1 who happened to have severe diarrhea or urinary tract infections, simultaneously. In our study, a male infant was found to have both urinary tract infection and PHA1. From our experience, imaging of the kidneys and adrenals, urinary analysis and tests for steroid precursors are recommended to exclude renal and adrenal causes. If the electrolyte disturbance cannot be corrected with the proper treatment towards primary disease in a relatively short time, molecular genetic analysis can be great helpful to make a correct diagnosis.
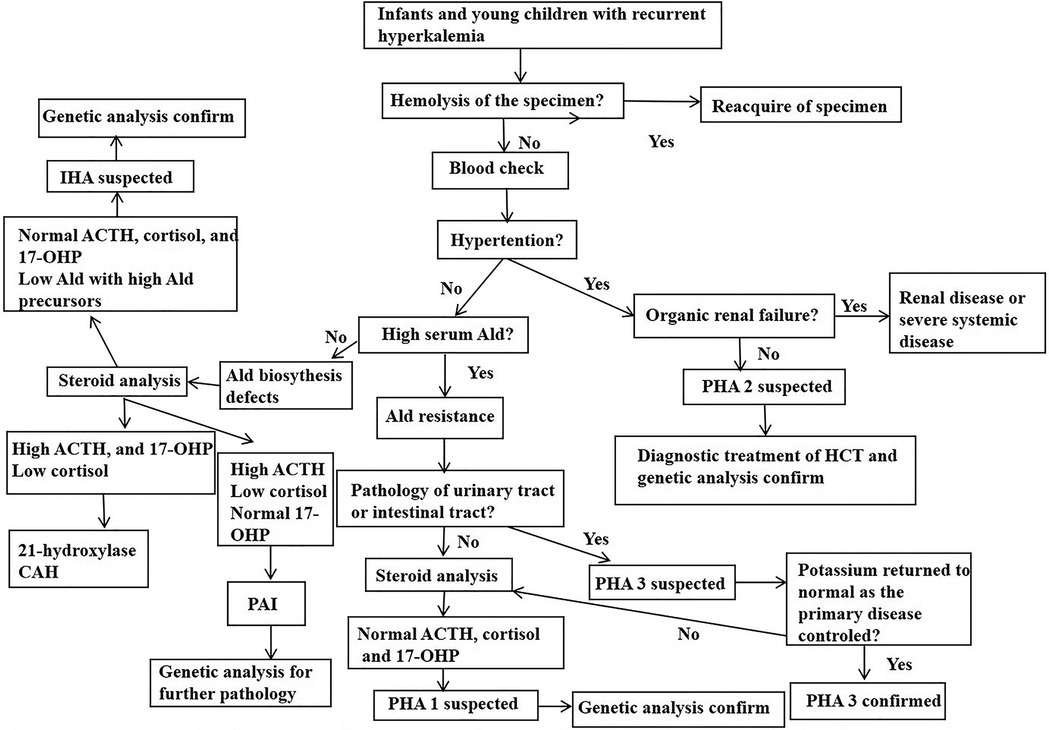
CAH, PAI, IHA and PHA may all present with similar clinical symptoms which can make these conditions hard for a specific diagnosis. This study proposes a diagnostic procedure for infants and young children with hyperkalemia who are suspected with Ald signaling defects. This may be helpful for physicians to make a quick and correct diagnosis (Figure 1).
Figure 1. Proposed diagnostic procedure for inants and young children presenting with hyperkalemia who is suspected of aldosterone signal defect. Ald, aldoterone; CAH, congenital adrenal hyperplasia; PHA, pseudohypoaldosteronism; IHA, isolated hypoaldosterone; HCT, hydrochlorothiazide; 17-OHP, 17-hydroxyprogesterone.
In this retrospective study, we also identified three novel heterozygous mutations of the KLHL3 and CUL3 genes which were associated with PHA2. Of these, there were two cases due to the KLHL3 gene: c.1484C > G (p.T495R) and c.470_471delinsTG (p.A157V). The third was a de novo mutation of c.560G > A (p.C187Y) in the CUL3 gene (Table 2).
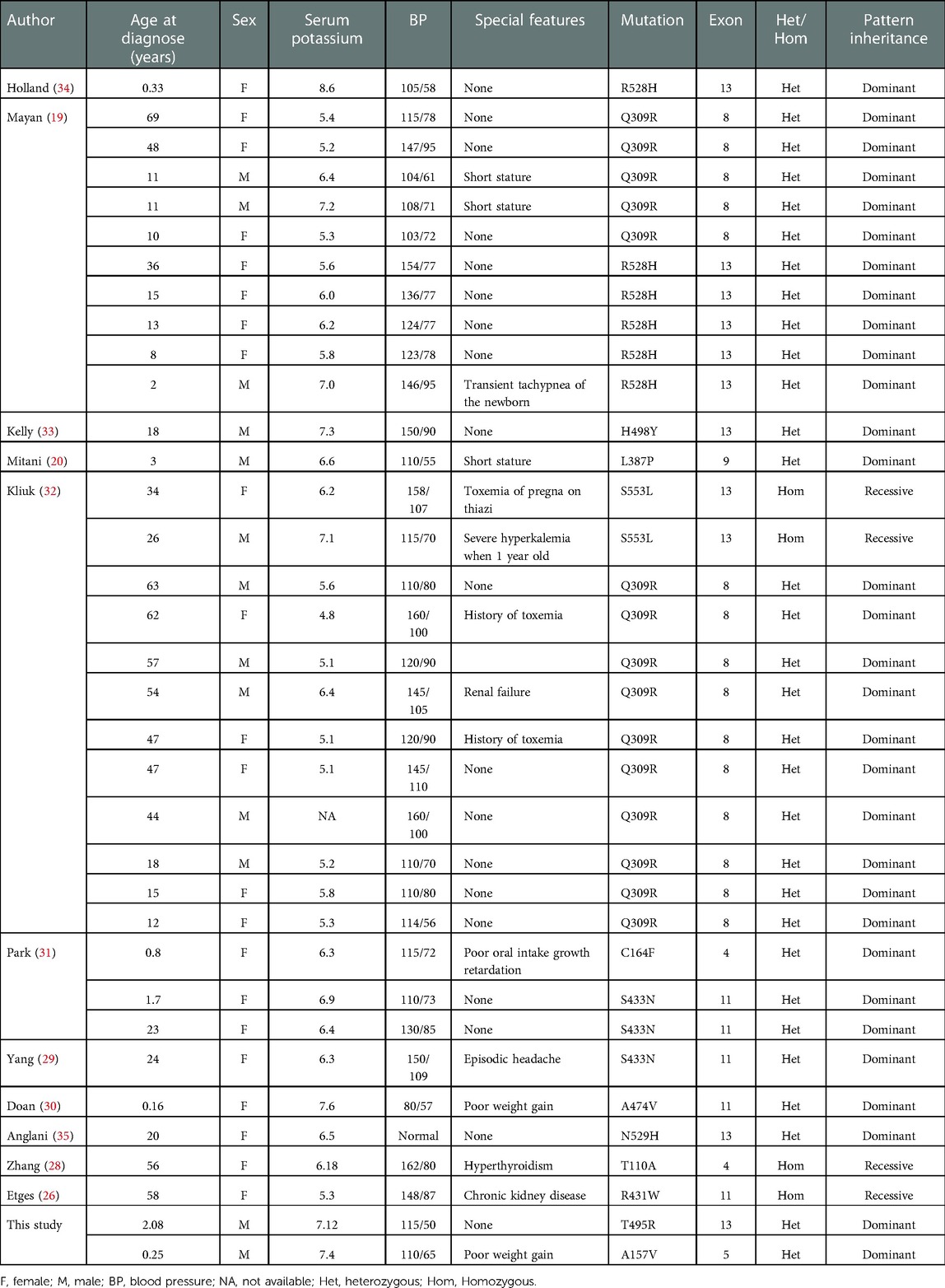
The KLHL3 protein is thought to have three domains: a BACK (BTB and C-terminal Kelch) domain, a N-terminal BTB domain, and Kelch-like repeats forming a six-bladed β-propeller structure (12, 14, 25). The mutation of p.A157V is located in the BTB domain, which plays an important role in binding to the CUL3 protein. The p.T495R residue is located in the fifth Kelch motif of the protein and is involved in substrate binding. The clinical and genetic features of patients with mutations in KLHL3 (19, 20, 26–34), including the present study and those who had clinical details published in previous reports, are summarized in Table 3. Heterozygous mutations are more common than homozygous ones. The age of the patients ranged from early infants to 69 years old. All of them had typical clinical symptoms including hyperkalemia and hypertension except for one 20-year-old female who had normal blood pressure. The mechanisms of the phenotype and genotype are still unclear. In our study, the two affected boys showed typical PHA2 presentation, but their 32- and 28-year-old father and mother both had the same KLHL3 mutation, respectively, but were asymptomatic and had no hyperkalemia. More research and clinical details are needed to accurately and convincingly describe the genotype-phenotype correlation in this condition.
Table 3. Review of the characteristics of PHA2 patients with KLHL3 gene mutations.
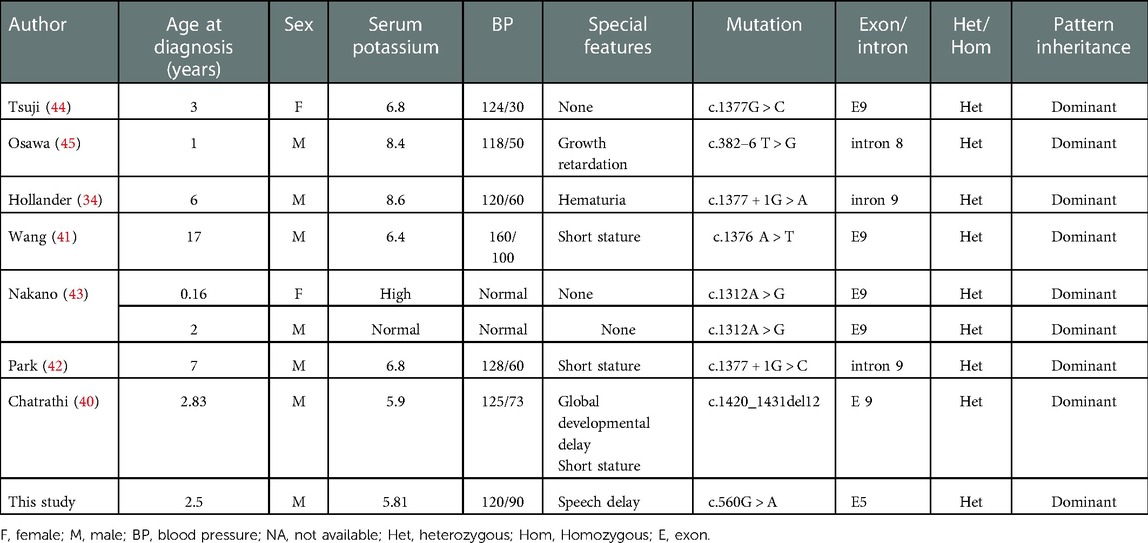
The CUL3 gene is located in 2q36.2 and is also a causative gene for PHA2. To date, the reported PHA2-related CUL3 variants are all clustered in introns 8 and 9 and exon 9. These resulted in an in-frame 57 amino acid deletion of exon 9 (residues 403–459) which produced a truncated form of the CUL3 protein (14, 36). However, we found a young child who presented with typical symptoms of PHA2 carrying a de novo mutation of CUL3 in exon 5. This mutation was predicted to be pathogenic by bioinformatic tools including PolyPhen2, SIFT, Mutation Taster and ClinPred. According to the American College of Medical Genetics and Genomics guidelines (37), the de novo variant was classified as likely pathogenic. Remarkably, the CUL3 gene coding protein, is a component of a cullin–RING E3 ubiquitin ligase (38). It is involved in a wide range of critical cellular processes, including targeting proteins for proteasomal degradation, protein activity modulation, interaction and localization (14). CUL3 mutations may affect these activities, and therefore, would produce a very broad range of phenotypes. Recently, a CUL3 novel mutation was found in patients with autism (39). As with our study, the young child with the CUL3 gene mutation suffered from development delay and speech dyspraxia. The same condition was reported by Chatrathi et al. (40). We further summarized the patients with PHA2 due to the CUL3 gene mutations (34, 40–45) from the literature (Table 4).
Table 4. Review of the characteristics of PHA2 patients with CUL3 gene mutations.
Correct therapy aiming at the cause specific of a disease is needed to treat patients. For patients with PAI, glucocorticoids combined with fludrocortisone (FC) could get a good curative effect. Treatment with FC and oral sodium supplementation are essential for patients with mineralocorticoid hormone deficiency, especially when they are at a young age. Thiazide diuretics is an effective treatment for patients with PHA2. All patients with PHA2 in our study had normal blood pressure as well as normalized blood electrolyte when hydrochlorothiazide was administered as the therapy.
ConclusionsIn this retrospective study, we describe the etiologies of Ald signaling defects seen in our hospital in infants and young children who presented with hyperkalemia and we subsequently proposed a diagnostic procedure for these patients. We also identified 3 novel mutations in the KLHL3 and CUL3 genes which were associated with PHA2. These cases broaden the clinical and genetic correlations towards PHA2. This is the first study to contain a complete PHA cohort. However, as a retrospective study it has a number of limitations. More prospective and mechanistic studies are required in this field. Nonetheless, this has provided additional valuable clinical experience for diagnosing and treating patients with hyperkalemia due to Ald signaling defects.
Data availability statementThe original contributions presented in the study are included in the article, further inquiries can be directed to the corresponding author.
Ethics statementThe studies involving human participants were reviewed and approved by Ethics Committee of the First Affiliated Hospital of Guangxi Medical University (2022-E356-01). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributionsDL and XL participated in the study design. XL and YSX drafted the manuscript and contributed equally. DL revised the manuscript. XL, YSX, JT, JZ and DZ participated in data collection and analysis. All authors contributed to the article and approved the submitted version.
FundingWe thank the Scientific research Foundation of the First Affiliated Hospital of Guangxi Medical University (grant no. 2010219) and the Guangxi Clinical Research Center of Pediatric Diseases (grant no: GUI KE AD22035219) for funding this project.
AcknowledgmentsWe are very grateful to the patient and families. The authors also thank DS of Imperial College London for editing the manuscript.
Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary materialThe Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2023.1092388/full#supplementary-material.
References1. Rossignol P, Legrand M, Kosiborod M, Hollenberg SM, Peacock WF, Emmett M, et al. Emergency management of severe hyperkalemia: guideline for best practice and opportunities for the future. Pharmacol Res. (2016) 113:585–91. doi: 10.1016/j.phrs.2016.09.039
PubMed Abstract | CrossRef Full Text | Google Scholar
4. Perry R, Kecha O, Paquette J, Huot C, Van Vliet G, Deal C. Primary adrenal insufficiency in children: twenty years experience at the sainte-justine hospital, Montreal. J Clin Endocrinol Metab. (2005) 90:3243–50. doi: 10.1210/jc.2004-0016
PubMed Abstract | CrossRef Full Text | Google Scholar
5. Turan I, Kotan LD, Tastan M, Gurbuz F, Topaloglu AK, Yuksel B. Molecular genetic studies in a case series of isolated hypoaldosteronism due to biosynthesis defects or aldosterone resistance. Clin Endocrinol (Oxf). (2018) 88:799–805. doi: 10.1111/cen.13603
PubMed Abstract | CrossRef Full Text | Google Scholar
6. Wijaya M, Ma H, Zhang J, Du M, Li Y, Chen Q, et al. Aldosterone signaling defect in young infants: single-center report and review. BMC Endocr Disord. (2021) 21:149. doi: 10.1186/s12902-021-00811-9
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Peter M, Bünger K, Drop SL, Sippell WG. Molecular genetic study in two patients with congenital hypoaldosteronism (types I and II) in relation to previously published hormonal studies. Eur J Endocrinol. (1998) 139:96–100. doi: 10.1530/eje.0.1390096
PubMed Abstract | CrossRef Full Text | Google Scholar
9. Akin L, Kurtoglu S, Kendirci M, Akin MA, Hartmann MF, Wudy SA. Hook effect: a pitfall leading to misdiagnosis of hypoaldosteronism in an infant with pseudohypoaldosteronism. Horm Res Paediatr. (2010) 74:72–5. doi: 10.1159/000281404
PubMed Abstract | CrossRef Full Text | Google Scholar
14. Boyden LM, Choi M, Choate KA, Nelson-Williams CJ, Farhi A, Toka HR, et al. Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities. Nature. (2012) 482:98–102. doi: 10.1038/nature10814
PubMed Abstract | CrossRef Full Text | Google Scholar
17. Martín-Rivada Á, Argente J, Martos-Moreno G. Aldosterone deficiency with a hormone profile mimicking pseudohypoaldosteronism. J Pediatr Endocrinol Metab. (2020) 33:1501–5. doi: 10.1515/jpem-2020-0239
CrossRef Full Text | Google Scholar
18. Li N, Li J, Ding Y, Yu T, Shen Y, Fu Q, et al. Novel mutations in the CYP11B2 gene causing aldosterone synthase deficiency. Mol Med Rep. (2016) 13(4):3127–32. doi: 10.3892/mmr.2016.4906
PubMed Abstract | CrossRef Full Text | Google Scholar
19. Mayan H, Carmon V, Oleinikov K, London S, Halevy R, Holtzman EJ, et al. Hypercalciuria in familial hyperkalemia and hypertension with KLHL3 mutations. Nephron. (2015) 130:59–65. doi: 10.1159/000381563
PubMed Abstract | CrossRef Full Text | Google Scholar
20. Mitani M, Furuichi M, Narumi S, Hasegawa T, Chiga M, Uchida S, et al. A patient with pseudohypoaldosteronism type II complicated by congenital hypopituitarism carrying a KLHL3 mutation. Clin Pediatr Endocrinol. (2016) 25:127–34. doi: 10.1297/cpe.25.127
PubMed Abstract | CrossRef Full Text | Google Scholar
22. Ou CY, Chen YJ, Lin GB, Chen MF, Chia ST. Case report: newborns with pseudohypoaldosteronism secondary to excessive gastrointestinal losses through high output stoma. Front Pediatr. (2021) 9:773246. doi: 10.3389/fped.2021.773246
PubMed Abstract | CrossRef Full Text | Google Scholar
23. Vantyghem MC, Hober C, Evrard A, Ghulam A, Lescut D, Racadot A, et al. Transient pseudo-hypoaldosteronism following resection of the ileum: normal level of lymphocytic aldosterone receptors outside the acute phase. J Endocrinol Invest. (1999) 22:122–7. doi: 10.1007/BF03350891
PubMed Abstract | CrossRef Full Text | Google Scholar
24. Moss S, Gordon D, Forsling ML, Peart WS, James VH, Roddis SA. Water and electrolyte composition of urine and ileal fluid and its relationship to renin and aldosterone during dietary sodium deprivation in patients with ileostomies. Clin Sci. (1981) 61:407–15. doi: 10.1042/cs0610407
CrossRef Full Text | Google Scholar
26. Etges A, Hellmig N, Walenda G, Haddad BG, Machtens JP, Morosan T, et al. A novel homozygous KLHL3 mutation as a cause of autosomal recessive pseudohypoaldosteronism type II diagnosed late in life. Nephron. (2022) 146:418–28. doi: 10.1159/000521626
PubMed Abstract | CrossRef Full Text | Google Scholar
27. Anglani F, Salviati L, Cassina M, Rigato M, Gobbi L, Calò LA. Genotype-phenotype correlation in Gordon's Syndrome: report of two cases carrying novel heterozygous mutations. J Nephrol. (2022) 35:859–62. doi: 10.1007/s40620-021-01083-1
PubMed Abstract | CrossRef Full Text | Google Scholar
28. Zhang R, Zhang S, Luo Y, Li M, Wen X, Cai X, et al. A case report of pseudohypoaldosteronism type II with a homozygous KLHL3 variant accompanied by hyperthyroidism. BMC Endocr Disord. (2021) 21:103. doi: 10.1186/s12902-021-00767-w
PubMed Abstract | CrossRef Full Text | Google Scholar
29. Yang Y, Ou Y, Ren Y, Tian H, Chen T. Hypertension accompanied by hyperaldosteronism, hyperkalemia, and hyperchloremic acidosis: a case report and literature review. Case Rep Endocrinol. (2020) 2020:1635413. doi: 10.1155/2020/1635413
PubMed Abstract | CrossRef Full Text | Google Scholar
31. Park JS, Park E, Hyun HS, Ahn YH, Kang HG, Ha IS, et al. Three cases of Gordon syndrome with dominant KLHL3 mutations. Pediatr Endocrinol Metab. (2017) 30:361–4. doi: 10.1515/jpem-2016-0309
CrossRef Full Text | Google Scholar
32. Kliuk-Ben Bassat O, Carmon V, Hanukoglu A, Ganon L, Massalha E, Holtzman EJ, et al. Familial hyperkalemia and hypertension (FHHt) and KLHL3: description of a family with a new recessive mutation (S553l) compared to a family with a dominant mutation, Q309R, with analysis of urinary sodium chloride cotransporter. Nephron. (2017) 137:77–84. doi: 10.1159/000475825
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Anglani F, Salviati L, Cassina M, Rigato M, Gobbi L, Calo LA. Genotype-phenotype correlation in Gordon's Syndrome: report of two cases carrying novel heterozygous mutations. J Nephrol. (2022) 35:859–62. doi: 10.1007/s40620-021-01083-1
PubMed Abstract | CrossRef Full Text | Google Scholar
36. Schumacher FR, Siew K, Zhang J, Johnson C, Wood N, Cleary SE, et al. Characterisation of the Cullin-3 mutation that causes a severe form of familial hypertension and hyperkalaemia. EMBO Mol Med. (2015) 7:1285–306. doi: 10.15252/emmm.201505444
PubMed Abstract | CrossRef Full Text | Google Scholar
37. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
PubMed Abstract | CrossRef Full Text | Google Scholar
39. da Silva Montenegro EM, Costa CS, Campos G, Scliar M, de Almeida TF, Zachi EC, et al. Meta-Analyses support previous and novel autism candidate genes: outcomes of an unexplored Brazilian cohort. Autism Res. (2020) 13:199–206. doi: 10.1002/aur.2238
PubMed Abstract | CrossRef Full Text | Google Scholar
40. Chatrathi HE, Collins JC, Wolfe LA, Markello TC, Adams DR, Gahl WA, et al. Novel CUL3 variant causing familial hyperkalemic hypertension impairs regulation and function of ubiquitin ligase activity. Hypertension. (2022) 79:60–75. doi: 10.1161/HYPERTENSIONAHA.121.17624
PubMed Abstract | CrossRef Full Text | Google Scholar
41. Wang L, Nie M, Guo F, Tian Z, Guo X, Zhang S. A case of novel mutation of Cullin 3 gene in pseudohypoaldosteronism type II. J Hypertens. (2022) 40:1239–42. doi: 10.1097/HJH.0000000000003117
PubMed Abstract | CrossRef Full Text | Google Scholar
42. Park JH, Kim JH, Ahn YH, Kang HG, Ha IS, Cheong HI. Gordon syndrome caused by a CUL3 mutation in a patient with short stature in Korea: a case report. J Pediatr Endocrinol Metab. (2022) 35:253–7. doi: 10.1515/jpem-2021-0361
PubMed Abstract | CrossRef Full Text | Google Scholar
43. Nakano K, Kubota Y, Mori T, Chiga M, Mori T, Sonoda S, et al. Familial cases of pseudohypoaldosteronism type II harboring a novel mutation in the Cullin 3 gene. Nephrology. (2020) 25:818–21. doi: 10.1111/nep.13752
PubMed Abstract | CrossRef Full Text | Google Scholar
44. Tsuji S, Yamashita M, Unishi G, Takewa R, Kimata T, Isobe K, et al. A young child with pseudohypoaldosteronism type II by a mutation of Cullin 3. BMC Nephrol. (2013) 14:166. doi: 10.1186/1471-2369-14-166
PubMed Abstract | CrossRef Full Text | Google Scholar
45. Osawa M, Ogura Y, Isobe K, Uchida S, Nonoyama S, Kawaguchi H. CUL3 Gene analysis enables early intervention for pediatric pseudohypoaldosteronism type II in infancy. Pediatr Nephrol. (2013) 28:1881–4. doi: 10.1007/s00467-013-2496-6
留言 (0)