
記住我
MTT is a malignant peripheral nerve sheath tumor (MPNST) with rhabdomyoblastic differentiation. In 1938, Masson and Martin first described this compound tumor. They proposed that this tumor induces skeletal muscle differentiation in the same way as normal nerves (1). It got the name as malignant salamander tumor in 1973, basing on previous observations that if the sciatic nerve was implanted into the dorsal side, the salamander would produce redundant limbs composed of striated muscle, bone and nerve tissue. They mimic other brain tumors on MRI, which brings challenges to preoperative diagnosis. This the first MTT case reported with concomitant pituitary adenoma in the sellar area.
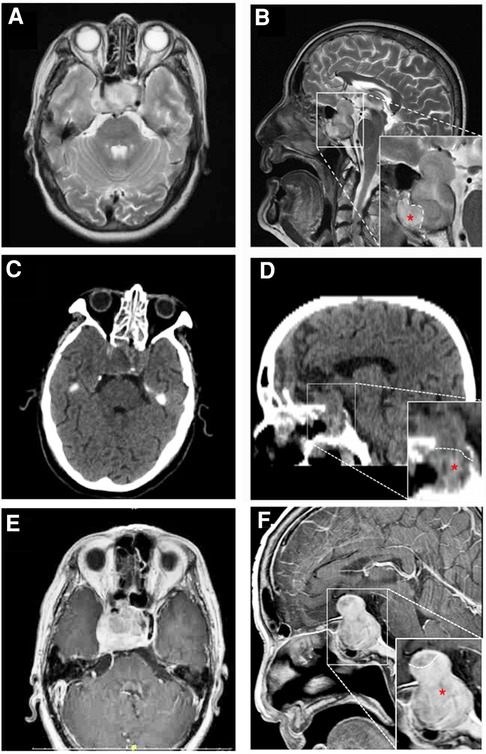
Case report History and presentationA 71-year-old man was referred to our hospital with impaired vision and intermittent nausea and vomiting in August 2020. He had a history of nausea and vomiting in June 2010. MRI revealed a lesion in the sellar region in a local hospital. At that time, he immediately underwent gross total surgical resection and pathological examination confirmed the lesion as a pituitary adenoma. Subsequently, he underwent adjuvant gamma knife treatment in September 2010. Thereafter, the symptoms disappeared, except that he experienced hyponatremia several times without formal treatment. In July 2020, the patient experienced vision loss, recurred intermittent nausea, and vomiting. The local hospital diagnosed cataract and a cataract surgery was performed accordingly. After the cataract surgery, the vision recovery was not ideal and continued to worsen. Meanwhile, intermittent nausea and vomiting did not alleviate. Intracranial MRI revealed an irregular mass with 2 distinctive components in sellar area (Figures 1A,B). An endocrine panel demonstrated an elevated serum follicle-stimulating hormone (FSH) level. Combined with medical history, it was considered that the tumor recurred. The patient was then referred to our hospital and underwent partial resection of the lesion with a transnasal transsphenoidal approach.
Figure 1. MRI (A,B,E,F) and CT (C,D) findings in a patient with pituitary adenoma (PA) and concurrent malignant triton tumor (MTT) in sellar area. On 20 days before the operation, axial image (A) and saggittal (B) T2-weighted images revealed that the lesion is consisted of two distinctive components with a clear border. The upper and lower components were histologically diagnosed as pituitary adenoma and MTT (red asterisk), respectively. Four days after the operation, axial (C) and saggittal (D) CT images showed that MTT component was totally removed, while residual PA could be seen in suprasellar area. Fifty-six days after the operation, axial (E) and saggittal (F) contrast-enhanced MR images revealed that MTT has recurred.
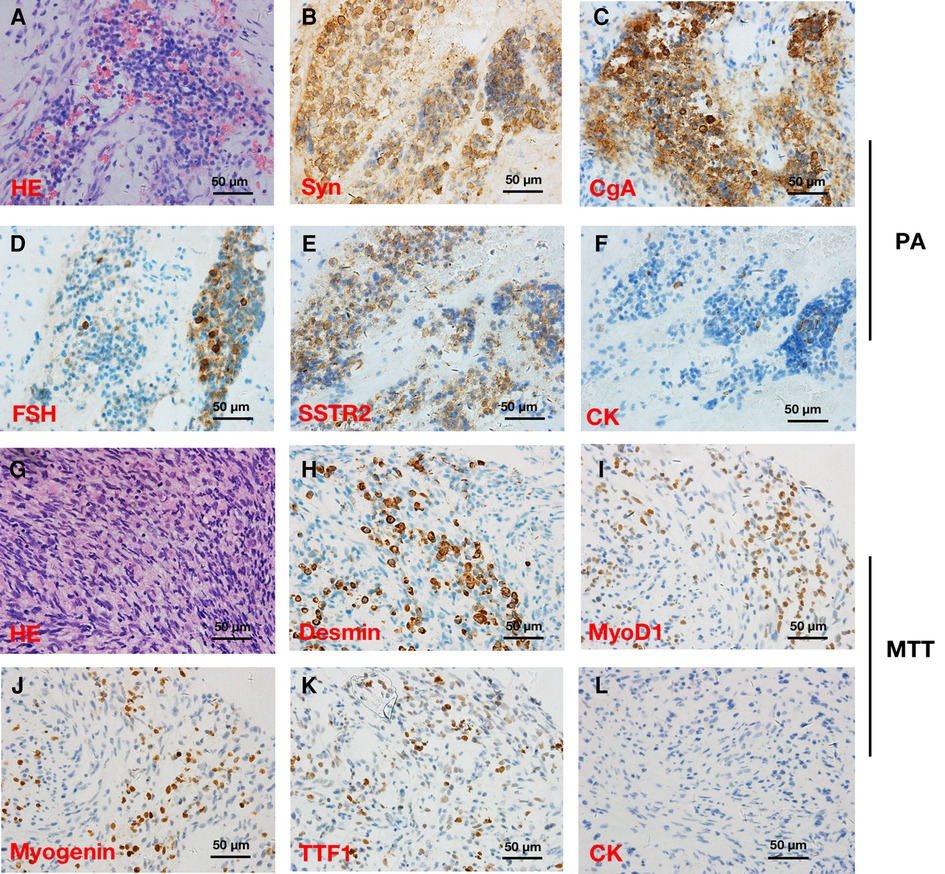
Pathological findings revealed that the tumor was composed of a pituitary adenoma (Figures 2A–F) in the upper area and a concurrent MTT in the lower area with a clear border (Figures 2G–L). In the pituitary adenoma component, immunostaining was positive for synaptophysin (Syn), chromogranin A (CgA), FSH, cytokeratin (CK), and somatostatin receptor 2 (SSTR2). In the MTT component, immunostaining was positive for Desmin, Myogenin, thyroid transcription factor-1 (TTF-1), and myogenin differentiation 1 (MyoD1). Interestingly, TTF-1, which could be found in 66.7% of schwannomas (2) and is also a novel marker of pituitary tumors of the posterior lobe (3, 4), was immunoreactive (Figures 2H). CK was immunonegative in the MTT portion. In addition, the average Ki-67 staining (MIB labeling index) in the PA and TT portions were 3% and 40%, respectively.
Figure 2. Hematoxylin and eosin (HE) and immunohistochemical stainings in a patient with pituitary adenoma (PA) (A–F) and concurrent intracraninal malignant triton tumor (MTT) (×400) (G–L). HE (A) and immunoreactivity of synaptophysin (Syn) (B), chromogranin A (CgA) (C), follicle-stimulating hormone (FSH) (D), somatostatin receptor 2 (SSTR2) (E), and cytokeratin (CK) (F) supported the diagnosis of PA. Round and fussiform cells with eosinophilic cytoplasm morphologically consistent with rhabdoid differentiation were observed in HE staining (G). Desmin (H), Myogenin (I), and myogenic differentiation 1 (MyoD1) (J) immunoreactivity supported the diagnosis of a MTT. Notably, TTF-1 (K) was immunopositive, while CK (L) was immunonegative in the MTT.
On the fourth day after the operation, computer tomography (CT) examination of the brain revealed that the tumor mass containing MTT component in sphenoidal sinus was gross-totally resected, while the pituitary adenoma was partially resected with residual tumor in the suprasellar area (Figures 1C,D). Post-operatively, the patient developed hyponatremia and recovered after active treatment. The patient's vision loss also recovered well after the operation. However, blurred vision recurred and continued to worsen 7 days after the operation. A re-examination of cranial MRI 56 days after the operation showed that the residual tumor had grown again (Figures 1E,F). The patient was blind and refused to undergo further surgical treatment and radiotherapy. Eventually, he died 64 days after the second operation.
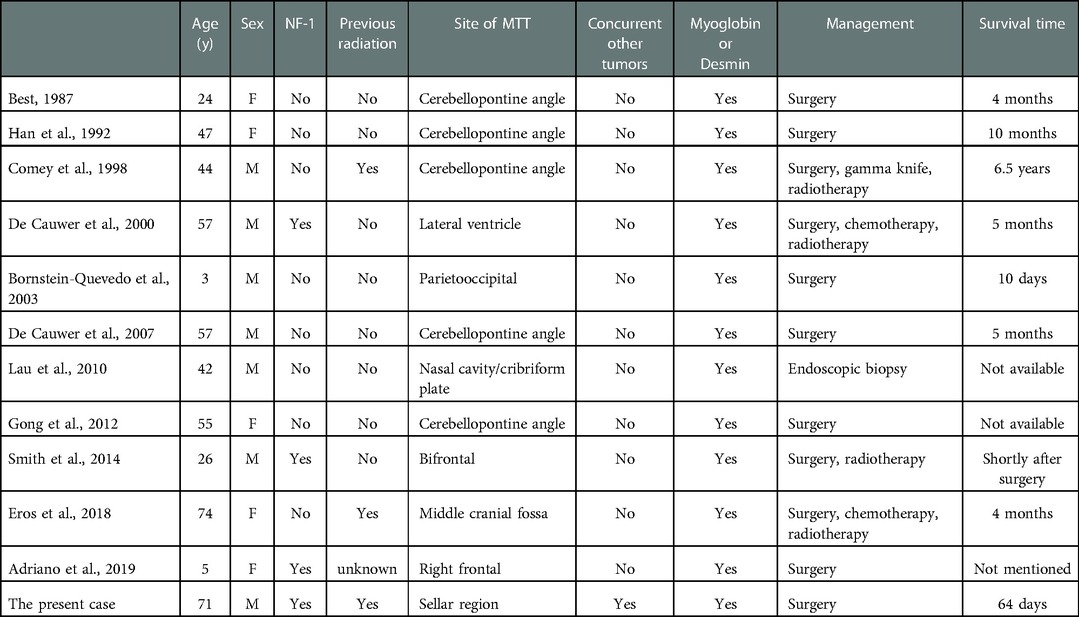
DiscussionMTTs is a subgroup of malignant peripheral nerve sheath tumors (MPNST), which are reported to exhibit rhabdomyosarcoma-like differentiation and follow a particularly aggressive course (5). Due to the rarity of MTT, merely eleven intracranial cases have been reported, which is summarized in Table 1. To the best of our knowledge, the present case is the first report of MTT in sellar region with a clear history of radiation and a concurrent relapsed pituitary adenoma.
Table 1. Summary of reports of intracranial malignant triton tumors (MTTs).
It is generally believed that the striated muscle cells in tumors are induced by or directly transformed from Schwann cells (6). In the present case, the visual acuity of the patient recovered completely after operation and the MTT component located apart from the optic nerve, which can basically exclude the origination of MTT from the optic nerve sheath.
MTTs mimic gliomas on MRI (7), where both of them show irregular masses, fuzzy boundaries, a peripheral increase, a low or equal signal on T1, and a high signal on T2 images (8). Genomic and proteomic studies have begun to reveal the complexity of chromosome and transcriptional changes behind these tumors, but reliable cytogenetic markers have not been established to distinguish MTT and MPNST and/or explain the obvious differences in their behavior (9–11). Daimaru et al. proposed a relatively widely accepted definition, including the following (12): (1) The first criterion is that, it is shown as a malignant schwannoma under the microscope and contains focal rhabdomyoblasts; The second criterion is that, the tumor is mainly composed of striated muscle and focal Schwann cells, which are found in neuropathy or NF-1 expression. Now, the diagnosis of MTT commonly depended on these criteria and on immunohistochemical findings (13).
The differential diagnosis of MTT includes other spindle cell malignant tumors, such as fibrosarcoma, synovial monophase sarcoma, leiomyosarcoma, low-grade fibroblast sarcoma, especially spindle cell rhabdomyosarcoma (14). Low-grade myofibroblastic sarcomas, synovial sarcomas, and fibrosarcomas do not contain rhabdomyoblasts and do not respond to skeletal muscle staining such as Desmin. Rhabdomyosarcoma, especially embryonic rhabdomyosarcoma, is mainly consisted of undifferentiated round cells and spindle cells (15). However, the composition of rhabdomyosarcoma is different from that of MTT, and the mean age of patients with rhabdomyosarcoma is younger than the patients with MTTs. In addition, there are usually high-density cell areas surrounding blood vessels, alternating with parvicellular areas rich in mucosal intercellular material.
In the present case, immunostainings were positive for skeletal muscle markers including Myogenin, Desmin, and Myod1, which supports the diagnosis. Interestingly, TTF-1 immunoreactivity was observed in the MTT component. Given that TTF-1 could be found in 66.7% of schwannomas (2), this result also supported the diagnosis of MTT. Moreover, in the 2017 WHO classification of pituitary tumors (4), TTF-1 expressing pituitary tumors of the posterior lobe represent a morphological spectrum of a single nosological entity. Considering the location of the tumor in our present case, we boldly speculate that there is possibility that MTT portion might arise from the mesodermal components in the posterior lobe of pituitary gland. Additionally, S100 protein is immunopositive in 50%–90% of MPNSTs (13), while S100 protein was negative in our case. We hypothesize that it might be explained by the following reasons: (1) the MTT tissue in our present case was not fully differentiated to express S100; (2) the S100-positive tissue was discarded or vacuumed during the surgical operation and was not provided for pathological examination.
It is reported that 8% of patients with MTT had been exposed to radiation, which is recognized as a risk factor (16–18). In addition, even therapeutic radiation can trigger the formation of MTT (19). Our report also provides evidence that the occurrence of MTT might be induced by radiation therapy.
Since MTT is rare and lack of prospective clinical trials, there is no standard treatment available. The 5-year survival rate of patients with MTT was 11%–14%, which was significantly lower than those patients with MPNST (34%–52%) (13, 14, 16). The adverse prognostic factors of MTTS mainly include NF-1, age, size, surgical method, positive surgical margin, early local recurrence, and the location of the tumor in the trunk (19).
Complete resection can improve the prognosis, reduce the risk of local recurrence and metastasis, and improve the efficacy of adjuvant therapy. In contrast, subtotal cytoreductive surgery cannot improve the survival rate of MTT patients (13). Adjuvant radiation therapy reduces the risk of death, but is not associated with a reduction in progression or recurrence (16). Although adjuvant chemotherapy has not been proved to be effective (16), there are certain evidence that patients who respond to neoadjuvant chemotherapy should also receive adjuvant therapy immediately (20). However, most MTT patients usually die within a few months, even after receiving optimal treatments (18).
In the future, we hope to have more cases and studies to better understand this tumor and provide more reliable diagnostic methods, especially pathological diagnostic criteria and more effective treatment strategies.
ConclusionIn summary, to our knowledge, this is the first report of MTT in sellar region with concurrent other tumors. This case had a history of radiation exposure after the first operation, which suggests that the occurrence of MTT might be caused by radiation. The patient received partial resection of the lesion with a transnasal transsphenoidal approach. The patient did not receive any adjuvant treatment postoperatively. Eventually, he died 64 days after the operation. Unfortunately, due to the rapid progress of the patient's condition, this case did not have much value for reference in treatment. In the future, we hope to have more cases and studies to better understand this tumor and provide more reliable diagnostic methods, especially pathological diagnostic criteria and more effective treatment strategies.
Data availability statementThe original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Ethics statementThe studies involving human participants were reviewed and approved by the Research Ethics Committee of Shandong Provincial Hospital Affiliated to Shandong First Medical University. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributionsJW and ZY analyzed and interpreted the data, and wrote the manuscript. SX analyzed and interpreted the data, and revised the manuscript. BL designed and conceptualized the study, interpreted the data, and revised the manuscript. All authors contributed to the article and approved the submitted version.
FundingThis study was supported by National Natural Science Foundation of China (no. 81902531).
AcknowledgmentsThe authors would like to thank our patient for allowing for his case to be presented.
Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References1. Weiss SW, Goldblum JR. Malignant tumors of peripheral nerves. In: Weiss SW, Goldblum JR, editors. Enzinger and weiss’s soft tissue tumors St. Louis: Mosby. (2008).
2. Wang DZ, Liu P, Yao L, Hao YH, Zhu RJ, Zhang T, et al. Aberrant expression of thyroid transcription factor-1 in schwannomas. Hum Pathol. (2018) 71:84–90. doi: 10.1016/j.humpath.2017.10.025
PubMed Abstract | CrossRef Full Text | Google Scholar
3. Hussein SE, Vincentelli C. Pituicytoma: review of commonalities and distinguishing features among TTF-1 positive tumors of the central nervous system. Ann Diagn Pathol. (2017) 29:57–61. doi: 10.1016/j.anndiagpath.2017.05.004
PubMed Abstract | CrossRef Full Text | Google Scholar
5. Ducatman BS, Scheithauer BW, Piepgras DG, Reiman HM, Ilstrup DM. Malignant peripheral nerve sheath tumors. A clinicopathologic study of 120 cases. Cancer. (1986) 57(10):2006–21. doi: 10.1002/1097-0142(19860515)57:10%3C2006::AID-CNCR2820571022%3E3.0.CO;2-6
PubMed Abstract | CrossRef Full Text | Google Scholar
6. Bornstein-Quevedo L, Peralta-Olvera F, Marhx-Bracho A, Rodriguez-Jurado R, De Leon-Bojorge B. Cerebral malignant nerve sheath tumor, triton tumor variant: case report. Pediatr Dev Pathol. (2003) 6(2):168–72. doi: 10.1007/s10024-001-0125-z
PubMed Abstract | CrossRef Full Text | Google Scholar
8. De Cauwer H, Bogers JP, Duwel V, den Hauwe V, Croese P, Van Marck E. An intracerebral intraparenchymatous triton tumor in a man with neurofibromatosis. J Neurol. (2007) 254(8):1009–11. doi: 10.1007/s00415-006-0469-4
PubMed Abstract | CrossRef Full Text | Google Scholar
9. Grobmyer SR, Reith JD, Shahlaee A, Bush CH, Hochwald SN. Malignant peripheral nerve sheath tumor: molecular pathogenesis and current management considerations. J Surg Oncol. (2008) 97(4):340–9. doi: 10.1002/jso.20971
PubMed Abstract | CrossRef Full Text | Google Scholar
10. Velagaleti GV, Miettinen M, Gatalica Z. Malignant peripheral nerve sheath tumor with rhabdomyoblastic differentiation (malignant triton tumor) with balanced t(7;9)(q11.2;p24) and unbalanced translocation der(16)t(1;16)(q23;q13). Cancer Genet Cytogenet. (2004) 149(1):23–7. doi: 10.1016/S0165-4608(03)00278-4
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Bridge RS Jr, Bridge JA, Neff JR, Naumann S, Althof P, Bruch LA. Recurrent chromosomal imbalances and structurally abnormal breakpoints within complex karyotypes of malignant peripheral nerve sheath tumour and malignant triton tumour: a cytogenetic and molecular cytogenetic study. J Clin Pathol. (2004) 57(11):1172–8. doi: 10.1136/jcp.2004.019026
PubMed Abstract | CrossRef Full Text | Google Scholar
12. Daimaru Y, Hashimoto H, Enjoji M. Malignant “triton” tumors: a clinicopathologic and immunohistochemical study of nine cases. Hum Pathol. (1984) 15(8):768–78. doi: 10.1016/S0046-8177(84)80169-0
PubMed Abstract | CrossRef Full Text | Google Scholar
13. Stasik CJ, Tawfik O. Malignant peripheral nerve sheath tumor with rhabdomyosarcomatous differentiation (malignant triton tumor). Arch Pathol Lab Med. (2006) 130(12):1878–81. doi: 10.5858/2006-130-1878-MPNSTW
PubMed Abstract | CrossRef Full Text | Google Scholar
14. Mentzel T, Kuhnen C. Spindle cell rhabdomyosarcoma in adults: clinicopathological and immunohistochemical analysis of seven new cases. Virchows Arch. (2006) 449(5):554–60. doi: 10.1007/s00428-006-0284-4
PubMed Abstract | CrossRef Full Text | Google Scholar
15. Leiner J, Loarer FL. The current landscape of rhabdomyosarcomas: an update. Virchows Arch. (2020) 476(1):97–108. doi: 10.1007/s00428-019-02676-9
CrossRef Full Text | Google Scholar
16. McConnell YJ, Giacomantonio CA. Malignant triton tumors–complete surgical resection and adjuvant radiotherapy associated with improved survival. J Surg Oncol. (2012) 106(1):51–6. doi: 10.1002/jso.23042
PubMed Abstract | CrossRef Full Text | Google Scholar
18. Kamran SC, Howard SA, Shinagare AB, Krajewski KM, Jagannathan JP, Hornick JL, et al. Malignant peripheral nerve sheath tumors: prognostic impact of rhabdomyoblastic differentiation (malignant triton tumors), neurofibromatosis 1 status and location. Eur J Surg Oncol. (2013) 39(1):46–52. doi: 10.1016/j.ejso.2012.09.001
PubMed Abstract | CrossRef Full Text | Google Scholar
19. Comey CH, McLaughlin MR, Jho HD, Martinez AJ, Lunsford LD. Death from a malignant cerebellopontine angle triton tumor despite stereotactic radiosurgery. Case report. J Neurosurg. (1998) 89(4):653–8. doi: 10.3171/jns.1998.89.4.0653
PubMed Abstract | CrossRef Full Text | Google Scholar
20. Thoennissen NH, Schliemann C, Brunnberg U, Schmidt E, Staebler A, Stegger L, et al. Chemotherapy in metastatic malignant triton tumor: report on two cases. Oncol Rep. (2007) 18(4):763–7. doi: 10.3892/or.18.4.763
留言 (0)