
記住我




For the initial phase of the project, the identification of the status of each pending application proved to be a success as it allowed for better coordination and management of applications. In addition, obtaining the status of the finalised products from each unit provided a list of applications that each unit can focus on (Fig. 3). Although allocation was conducted at the same time by the Health Products Authorisation (HPA) section, the units did not initiate the evaluations at the same time. With the improved process this would be alleviated as communication to the applicant will be synchronised for all the applications.
New Applications—Risk-Based AssessmentsThe planning of Phase 2 of the 2015 backlog involved engagements with other stakeholders for the success of the project. The stakeholders, such as the applicants and the Expert Committees, held a wealth of knowledge regarding processes, historical information, industry insight, and in the planning and execution of the project for new applications. It was therefore imperative that they were consulted in the decision-making of the project to allow for a seamless process to occur. The proposed process was outlined, and modifications were made where necessary until a consensus was reached to initiate the pilot project.
The proposed process was communicated with all stakeholders involved, which included the CEOs of the pharmaceutical companies in the pilot study, the P&A Expert Committee Members and Unit, the Clinical Evaluations Expert Committee Members and Unit, the members of the MCC Registration Committee, and the Industry Technical Group (ITG). It was agreed that all new applications not yet reviewed should be resubmitted to facilitate review. This is because the submission for these products were between 2011 and 2012, thus the information in the dossiers was outdated. It was observed that the frequent recommendations for the old applications, since 5 years had lapsed, were on updates of the stability data, updated Certificate of Suitability (CEP), changes in the methods of synthesis, changes in the API manufacturers, changes in the FPP manufacturers, etc. This meant that several changes had occurred to a product over time, and in some instances, the product was considered non-existent as the final product manufacturers were no longer in business or were no longer manufacturing it. Thus, after registration, the applicant would apply for post-registration amendments, and by registering the products that essentially no longer exist, MCC was shifting the work to the Post-Registration Unit without eliminating the burden the authority faced. Hence, applicants were requested to uplift, update and re-submit the paper documents. Uplifting of the paper dossiers was conducted 2 months prior to the re-submission date, which gave applicants enough time to update their applications.
Consultation with the applicants resulted in withdrawal of 31% of the applications due to the lack of a business need for the product and only 99 master applications were left for the pilot study. The dossiers were re-submitted between 12 and 16 September 2016, distributed to the respective units, and evaluated by the PEM, P&A Pre-Registration Unit during the evaluation week held on 19–23 September 2016.
Even with the two phases as detailed above, by 2018 the backlog of applications had increased to 8,220. In 2018, the authority embarked on a project called the Backlog Clearance Programme aimed at clearing the existing backlog over a specified time. The planning and development of the project was initiated in February 2018 through the assistance of a project consulting firm, which assisted in the quantification of the backlog. Inherited processes and practices from the former MCC were re-assessed and the backlog project was initiated in August 2019 to support new methodologies required to achieve the goal of clearing the backlog of applications [7]. The project was initiated through the assistance of funding from government, development partners and donors [48].
The applicants were initially requested to indicate if they would like to include their applications in the Backlog Clearance Project. Upon analysis of the business need and proposed timeframe to submit there were 4,610 applications that opted out of the project and 99 applications were withdrawn. Not being part of the backlog project meant once the dossier was ready for resubmission with the new requirements, it would be submitted to the BAU section of SAHPRA. The in-process applications that were near finalisation, by either unit, were assessed in the BAU and concluded. Thus, SAHPRA initiated the Backlog Clearance Project in August 2019 with 3,343 applications, which translates to 1,364 master applications.
The Backlog Clearance Project utilised 56 external domestic and international evaluators to conduct the scientific assessments as well as the internal evaluators from the BAU section working overtime to assist with the project. By May 2021, 34% of the applications had been cleared. This was nearly 2 years after the initiation of the project where the intent was to eliminate the backlog in 2 years. The program was extended by 1 year and 5 months to December 2022 and the delay in the clearance was attributed to the assessments conducted within the PEM, P&A Pre-Registration component due to the bulk of the work being done in this unit [49]. Hence, the necessity for the refinement of the risk-based assessment in September 2021 in an effort to conclude the Backlog Clearance Project in the set time. The 63 applications that were next in line for allocation were in re-submission window eight (8) and were therefore used in the 2021 pilot study.
In 2019 when the backlog clearance programme was initiated, the business-as-usual (BAU) section was provided with the opportunity to start on a clean slate while the backlog clearance programme dealt with all the ~8,220 applications. In the period 2019 to 2022, SAHPRA amended its processes and put systems in place such as the inclusion of a tracker that allows all units to monitor each other; however, even with that, a backlog formed within the BAU section of SAHPRA. The tracker was aimed at providing transparency and synchronisation within the units; however, this did not correct the misalignment as units could still allocate the same applications at different times and communicate the queries at different times. The solution to this would have been to have one set of queries from the different units communicated at the same time by the PC, as conducted in the 2015 study to ensure alignment within units at all times. This meant some units would finalise applications before others, which would lead to misalignment. It should be noted that the root cause of the backlog was not as a result of one factor such as the misalignment of units only, there are a number of reasons, which are detailed in the study, and which is why the risk-based assessment approach was developed as an end-to-end registration process providing corrective or preventative measures or solutions to prevent the root causes from occurring in future.
Risk-Based Assessment ProcessRegistration ProcessA reassessment of processes was necessary for the authority for improved efficiencies. An improved registration process was employed as detailed in Fig. 4.
The following were improved in the developed process illustrated in Fig. 4:
Previously, the units were only allocated an application by the HPA, thereafter communication with the applicants would be made by the separate units. A Portfolio Coordinator (PC) responsible for coordinating and collating outcomes from the units was introduced as one communication to the applicants.
The introduction of the Inspectorate Unit confirming the Good Manufacturing Practice (GMP) status before allocation to other units was included since previously this would only occur once the scientific assessments had been concluded by the PEM, P&A and clinical evaluations of Pre-Registration Units. The inspections being conducted towards the end of the process would further delay the registration of applications.
The use of a risk-based approach to conduct scientific assessments to reduce the assessment times by the PEM, P&A Pre-Registration Unit with assessments focused on the critical quality attributes of the product.
The use of a pre-populated evaluation template to aid in the reduction of evaluation times. This allowed for the technical person to screen the applications to check if the updated information, such as the updated stability data, is as per the requested shelf-life, the updated Certificate of Suitability (CEP) is included, etc.
Frequent peer review meetings. For the 2016 pilot study, an evaluation week approach was used where a week was blocked for evaluation, during which towards the end of each day evaluators discussed the reports and query letters sent to the HPA. This promoted scientific knowledge sharing and ensured that queries going out to the applicants were critical aspects to be addressed in the dossier and that the queries were standardised. This was only conducted once, and the rest of the applications awaited the P&A Committee meetings held on a 6-weekly basis. This resulted in some delays.
In the refined process in 2021, weekly peer review meetings were introduced, which allowed for better throughput of query letters to the applicants. The selection of the date for each peer review session was based on the availability of evaluators using the When Available poll. The reports were then compiled into meeting documents and uploaded on Google Docs well in advance to allow evaluators to provide their comments. The living document would then show all comments in real-time, allowing all evaluators to see each other’s comments. This assisted in drastically reducing the meeting sessions as only specific points of discussion, highlighted by the peer-review panel, were discussed. Most other aspects were collaboratively deliberated on during the real-time discussions via Google Docs.
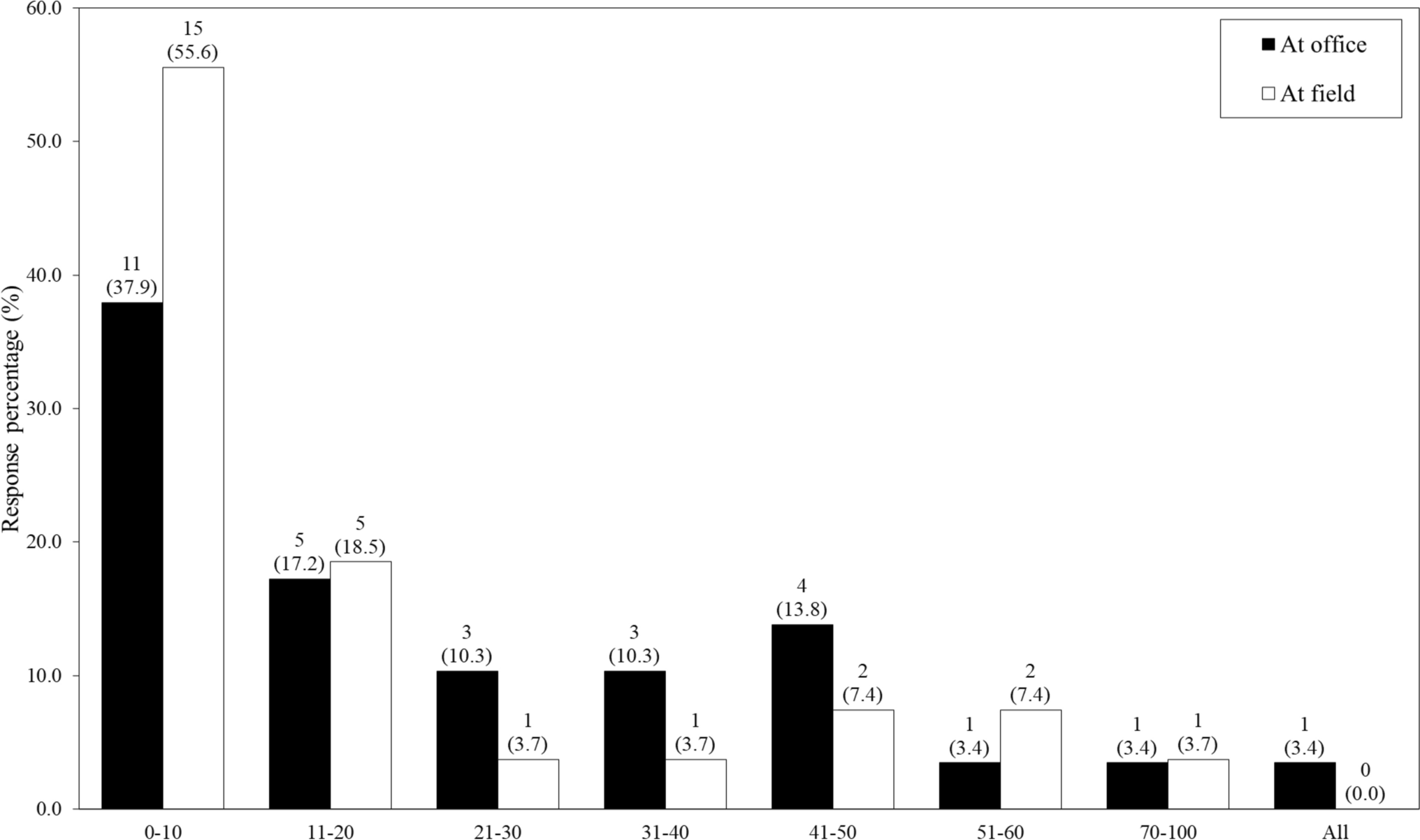
In the refined process this was further reduced to 10 working days; however, applicants could request an extension if required. The requests for extension were for 41% of the responses, therefore the response timeline was increased to 15 working days for initial responses and 10 working days for further responses.
Once this robust process had been concluded, the products were classified according to risk.
Risk ClassificationAhead of assessing the aspects of the API and FPP, prior work conducted by other NRAs or Regulatory Institutions should be considered. Recognition of the work previously done is termed as reliance. And, according to the WHO, reliance is defined as the act whereby one regulatory authority in one jurisdiction may consider and give significant weight to totally or partially rely upon scientific assessments or inspection reports performed by another authority or trusted institution in reaching its own decision [20]. The relying authority uses this work according to its own scientific knowledge and regulatory procedures and retains its own regulatory responsibilities. Historically, SAHPRA had not implemented this review pathway until 2019 when the backlog clearance programme was initiated [48]. The authorities with which SAHPRA aligns itself and uses the unredacted reports of are the European Medicines Agency (EMA); Health Canada; Medicines and Health Products Regulatory Agency (MHRA) in the United Kingdom; Ministry of Health, Labour and Welfare (MHLW) in Japan; Swiss Agency for Therapeutic Products (Swissmedic); Therapeutic Goods Administration (TGA), Australia; and the FDA [50]. SAHPRA is also currently utilising partial reliance through the use of submissions such as CEPs by the European Directorate for the Quality of Medicines (EDQM) and Certificates of Prequalification (CPQs) of the API by the World Health Organisation Prequalification Team: Medicines (WHO PQTm). The developed template in Table 2 therefore accommodates the reliance aspect as well during risk classification.
The non-reliance critical aspects are also considered during quality and efficacy (bioequivalence) aspects of products submitted for approval, and detailed below to assist in the overall classification of the product.
When it comes to defining the risk pertaining to the API, the following key aspects of the API are assessed:
Availability of a valid CEP/CPQ (Certificates of Prequalification (CPQs))
Pharmacopoeial status of the API
Biopharmaceutics Classification System (BCS) of the API (in particular aqueous solubility)
Solid state properties (solubility, hygroscopicity, particle size distribution (PSD) and polymorphism)
The concentration of the API in the FPP.
The key aspects to be considered in the FPP are:
Pharmacopoeial status of the FPP
Type of dosage form
Complexity of the manufacturing process
Excipients
Container closure system (CCS).
The key aspects in the bioequivalence study:
Based on the identified aspects to consider as stated in Table 2, a product could be classified as low or high risk.
Critical Areas to be Reviewed for Low-Risk ProductsA combination of literature reported by Tran et al. [32] and the concept paper by the WHO [19], as well as a wide array of expert advice garnered on the approach, categorically assisted in the determination of the critical attributes of manufacturing and overall risk ranking of the product. With this information, the CTD sections and extent of evaluation thereof could be established. The areas of concern have been included below and will be thoroughly evaluated for low-risk applications. The relevant templates are used for assessment with the critical sections included.
The identified critical sections of the CTD for low-risk applications are as follows:
Module 1.3 Labelling and packaging (Professional Information (PI), Patient Information Leaflet (PIL) and Label)
Module 1.7.4.1 Batch Release
Module 1.10 Foreign regulatory status
Module 3.2.S. Active Pharmaceutical Ingredient
3.2.S.1.3 Physico-chemical properties (depending on dosage form)
3.2.S.2.2 Method of synthesis (N/A if CEP/CPQ is submitted)
3.2.S.3.2 Impurities (N/A if CEP/CPQ is submitted)
3.2.S.4.1/2 Specifications (N/A if CEP/CPQ is submitted, however, assess the API specifications by the FPP manufacturer)
3.2.S.7 Stability (N/A if retest period is stipulated on CEP/CPQ)
Module 3.2.P Finished Pharmaceutical Product
3.2.P.1 Components and composition of the final product
3.2.P.3.3 Manufacturing process/Batch Manufacturing Record (BMR)
3.2.P.5.1 Specifications
3.2.P.7 Container closure system
3.2.P.8 Stability
Bioequivalence
The sections proposed for the bioequivalence section are included below and are in line with ICH and EMA requirements [51,52,53]. In the case where a BCS-based biowaiver is requested (BCS class I and III applications), only two sections would be assessed. These include the details of the test and reference product used in the study and comparative dissolution profiles, thus reducing the assessment review times. This template, used as an evaluation tool, would reduce the current reported evaluation timelines, as it is designed to point out and discuss critical aspects of the biostudy.
The identified sections from the bioequivalence template are as follows:
Details of the test and reference product used in the study (applicable for biowaiver request)
Comparative dissolution profiles (applicable for biowaiver request)
Study method and design
Summaries of statistical and pharmacokinetic data
Bioanalytical report parameters.
Certain sections are excluded from evaluation for low-risk applications. The rationale for these exclusions, which addresses the risk mitigation for each, are as follows:
Batch analyses (3.2.S.4.4 and 3.2.P.5.4) are not evaluated for low-risk applications because the stability results (3.2.S.7.3 and 3.2.P.8.3) at the initial time point essentially serve as batch analyses. In addition, the impurities section also includes profiling of the impurities and residual solvents formed, thus these sections mitigate the risk since they are assessed.
Reference materials sections (3.2.S.5 and 3.2.P.6) are for documentation purposes and do not need to be assessed since the API would have been confirmed already in preceding sections, such as the method of synthesis, impurity section and specifications. In most cases, 3.2.P.6 refers to section 3.2.S.5 of the dossier. The working standard and primary standards are those manufactured by the applicant and synthesis would, therefore, be in line with the proposed methods.
Pharmaceutical development (3.2.P.2) is not assessed for low-risk applications, because this is research and development conducted by the manufacturer for optimisation of the final manufacturing process for commercial product/s. The final proposed manufacturing process is then assessed in section 3.2.P.3.3 and the information is verified by the batch manufacturing records. In addition, for the oral solid dosage forms that require the submission of a bioequivalence study, certain critical aspects of the pharmaceutical development section are evaluated. These include in vitro dissolution studies as these are covered in the bioequivalence template for evaluation. For solid oral dosage forms, selection of inactive pharmaceutical ingredients (IPIs) is covered by the bioequivalence assessment where similarity to the reference product is reviewed, and in the case where the excipients are not similar to the reference product, API-excipient compatibility should be confirmed under 3.2.P.2. In the case of liquid dosage forms, excipient similarity to the reference is confirmed under Module 3.2.R.1.4.1 and in the case where the excipients are not similar to the reference product, API-excipient compatibility would be confirmed under 3.2.P.2. The designed templates therefore provide guidance for these.
Module 3.2.P.3.1 details the full name and address of the final product manufacturer. The name of the final product manufacturer is confirmed in the administrative table at the beginning of the pre-populated template. In addition, the Inspectorate Unit confirms and validates this during inspections.
Batch formula (3.2.P.3.2) is not assessed since it is confirmed during assessment of the batch manufacturing records, which consist of actual quantities of API/s and IPI/s used for the proposed batch(es).
Validation of analytical methods (3.2.S.4.3 and 3.2.P.5.3) is not assessed because the product would be pharmacopoeial and only verification is then required. In addition, specification limits provided found to be within ICH requirements will be confirmed since the specification section is assessed for low-risk applications. At most, the evaluator may only confirm the submission of the reports for noting for low-risk applications.
Critical Areas to be Reviewed for High-Risk ProductsIf a product is classified as high risk, additional sections over and above the ones identified for low risk would also require thorough evaluation and reporting on the respective templates. The additional sections to assess for high-risk products include the following:
Module 1.3 Labelling and packaging (PI, PIL and Label) – same as low-risk
Module 1.7 Good Manufacturing Practice – same as low-risk
Module 1.10 Foreign regulatory status – same as low-risk
Module 3.2.S Active Pharmaceutical Ingredient
Module 3.2.P Finished Pharmaceutical Product
3.2.P.2 Pharmaceutical development of the FPP
3.2.P.3.5 Process evaluation of the FPP validation
3.2.P.5.3 Validation of analytical methods for the FPP
3.2.P.7 Container closure system (for sterile applications)
Bioequivalence
Details of the test and reference product used in the study (applicable for biowaiver request)
Comparative dissolution profiles (applicable for biowaiver request)
Study method and design
Summaries of statistical and pharmacokinetic data
Bioanalytical report parameters
The justification stated above for the sections that are not to be assessed are also applicable for high-risk applications. Note that risk classification will not be applied to NCEs and biological applications; instead full review will be conducted due to the criticality of the medicines.
Summary of Results on the Risk-Based ApproachIn the second phase of the 2015 backlog pilot project for new applications, all 99 master applications were finalised within 9 months, with the median time calculated as 90 calendar days. The outliers were noted as 7, 8 and 9 months as indicated in Fig. 5. These were due to the FPP manufacturers receiving a negative status and therefore inspection had to be arranged by the Inspectorate Unit before evaluation could take place. There were other instances where the applicants requested an extension to submit responses, and this led to the delay in finalisation. For the refinement of the process in 2021, a median finalisation time of 68 calendar days was obtained (Fig. 5). Of the 63 applications, six were withdrawn while in-process in the response phase. However, the initial evaluation was already conducted for these so they were included in the calculations of evaluation times.
From the 63 applications, 21 applications were classified as high risk and 42 classified as low risk as depicted in Table 5. From Table 5, it is observed that all immediate-release tablets and capsules were low risk, which constitute 51% of the applications. From the 90% generic applications that SAHPRA receives, most of these are pharmacopoeial and well-known with readily available extensive research conducted on them; therefore, due to this, classification would be low risk. In addition, the dosage forms were not novel, therefore overall classification was low risk. The same applies for the other dosage forms classified as low risk.
Assessment TimelinesFigure 6 illustrates the reported evaluation times by the evaluators who were part of the two risk-based assessment pilot studies in 2016 and 2021. The graphical depiction shows the calculated median values as 6.3 and 7.0 h in 2016 and 2021, respectively, for low-risk quality assessment timelines. As observed from Table 4, products classified as low risk were immediate-release tablets and capsules, topical gels, mouth wash, throat spray, oral syrups and oral solutions. The median values for high-risk quality assessments were reported as 9.5 and 10 h from the two pilot studies, respectively. Products classified as high risk were sterile intravenous injections and infusions, ophthalmic solutions, delayed-release tablets and sterile lyophilised powders. The bioequivalence study assessment times were 8.4 and 8.0 h using the proposed template and biowaivers reported as 2.3 and 2.6 h with initial response assessment times as 2.6 and 3.4 h. The calculations above were based on a simplified submission that contains one API from one API manufacturer who submitted an Active Pharmaceutical Ingredient Master File (APIMF), with only one FPP manufacturer applied for. In a case where a CEP was submitted, the median evaluation times were 5–6 h for low-risk and 7–8 h for high-risk products; when two APIMFs were submitted, the evaluation times were 11–12 h for low-risk and 13–14 h for high-risk products. This resulted in the deduction that one APIMF assessment takes 4–5 h and one FPP takes 5–6 h to assess for high-risk applications. The reported medians have resulted in a reduction in the assessment times without the compromise to quality as only critical sections that will impact the quality of the product are adequately assessed.
For the Phase 2 pilot study conducted in 2022, the quality assessment timelines for high risk is reported as a median of 14 h and 10 h for low risk. The increased assessment timeline is due to the different quality template used that has been pre-populated by the applicant. The evaluators therefore would spend time validating the information populated by the applicant with the scientific information in the dossier to ensure that accurate information was completed.
Once applications that undergo the risk-based assessment pathway are registered, the following post-marketing surveillance or monitoring procedures were proposed and will be conducted:
The applicant will be requested to provide the Post-Registration reports on a yearly basis to Pharmacovigilance and annual product review report to the Inspectorate Unit. Depending on the information submitted on the reports, the Inspectorate could perform inspections of the non-compliant manufacturer/applicant.
Ongoing post-marketing surveillance will be conducted on the products by the Inspectorate Unit.
Re-evaluation of the information (dossiers) after 5 years will be conducted on all applications.
留言 (0)