
記住我
CD4 T cells promote a functional CD8 T-cell response, while CD4 T-cell deficiency drives increased CD8 T-cell exhaustion in settings of chronic antigen stimulation, like cancer. CD4 T cells are required for PD-1 blockade efficacy, while terminally exhausted CD8 T cells (Texterm) are unresponsive to PD-1 blockade. Glioblastoma is a demonstrably CD4-deficient tumor and unresponsive to PD-1 blockade therapy. The underlying basis for the resistance to PD-1 blockade in glioblastoma is unknown.
WHAT THIS STUDY ADDSOur results demonstrate that efficacy of PD-1 blockade therapy in brain tumors is reliant on a responsive subset of CD8 T cells maintained by functional CD4 T cells; however, a dysfunctional CD4 T-cell compartment may be circumvented with synergistic pharmacotherapies.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICYIntroductionThe implementation of checkpoint blockade therapy, an immunotherapy centered on reinvigorating an exhausted immunological response, has fundamentally altered oncology. However, clinical benefit has not yet been observed in glioblastoma.1 Although once considered an ‘immunologically privileged’ microenvironment, mounting data demonstrate active immune monitoring within the central nervous system (CNS), particularly in the diseased state with associated disruption of the blood–brain barrier.2 The presence of CNS immunosurveillance is underscored by the fact that metastatic disease to the brain responds to checkpoint therapy equivalently to extracranial disease.3 Therefore, the lack of response to checkpoint blockade therapy in glioblastoma is likely a disease-specific rather than a site-specific phenomenon, underscoring the need to better understand glioblastoma immunobiology.
PD-1 blockade relinquishes inhibitory signals and upregulates effector function of exhausted tumor-specific T cells, leading to restoration of antitumor immunity. Exhausted CD8 T cells (Tex) are a distinct T-cell lineage defined by the progressive loss of proliferative capacity and secretory effector cytokine production.4 5 Tex subsets were initially defined in mouse chronic viral infection models, in which dysfunction was correlated with progressive expression of inhibitory cell surface proteins such as PD-1, LAG3, and TIM-3.6 The application of single-cell technologies has provided additional granularity to the extensive heterogeneity of this lineage. Importantly, Tex subsets with differential responsiveness to checkpoint blockade have been characterized: PD-1 blockade-responsive progenitor exhausted CD8 T cells (Texprog) and unresponsive terminally exhausted CD8 T cells (Texterm).4 6–11 The Texprog population is defined by retained Tcf7 expression, critical to both maintain self-renewal and reactivate effector cytokine production.8 Conversely, in the Texterm subset, Tcf7 expression is lost, driving reduced proliferative potential, downregulated effector cytokines, and increased expression of coinhibitory immune checkpoint molecules through the high-mobility group box DNA-binding transcription factor, TOX. The progressive development of this Texterm state is driven by chronic antigen stimulation. Thus, one potential mechanism for glioblastoma resistance to PD-1 blockade therapy is a predominance of unresponsive Texterm.6 9 12
To evaluate this hypothesis, we studied two complementary murine glioblastoma models, GL261 and CT2A, with differential responsiveness to PD-1 blockade. While PD-1 blockade improves survival in mice with established intracranial GL261 tumors, PD-1 blockade is completely ineffective in mice bearing CT2A tumors.13 14 Despite the differences in sensitivity to PD-1 blockade therapy, these models share key similarities: (1) carcinogen induction with high mutational loads, (2) progressive growth when orthotopically transplanted into syngeneic C57BL/6 mice, and (3) well-defined Major Histocompatibility Complex (MHC) class I-restricted neoantigens that endogenously prime neoantigen-specific CD8 T cells. Thus, comparative analysis of these two models may provide insights into the underlying basis for lack of PD-1 blockade efficacy in brain tumors.
Prior studies have shown CD4 T-cell deficiency in both murine chronic viral infection models and HIV-positive patients leads to increased expression of exhaustion markers on CD8 T cells, suggesting CD4 T cells restrict the development of a more severe CD8 T-cell exhaustion phenotype.5 15 We have previously shown that there are significantly fewer CD4 T cells infiltrating CT2A tumors compared with GL261 tumors, and the CT2A-infiltrating CD4 T cells that are present are more dysfunctional than GL261-derived CD4 tumor-infiltrating lymphocytes (TILs).14 16 Similarly, when CD4 T cells are pharmacologically depleted from GL261 tumor-bearing mice, PD-1 blockade is ineffective.17 These observations suggest CT2A-depleted and CD4-depleted GL261 serve as useful models to evaluate the importance of CD4 T cells on CD8 T-cell exhaustion, which has direct relevance to the CD4-deficient state observed in patients with glioblastoma.
We used single-cell RNA sequencing (scRNA-seq) to analyze the tumor-infiltrating T cells of these three tumor models and observed a gradient of Tex that closely resembles previously described Texterm and Texprog subsets. Furthermore, we demonstrate that the PD-1 blockade-responsive GL261 model is enriched for Texprog cells, while the PD-1 blockade-unresponsive CT2A model is predominantly composed of Texterm cells. Consistent with the previously described role of CD4 T-cell loss in driving more severe CD8 T-cell exhaustion in mouse models and human disease, we note that CD4 T-cell depletion is associated with an abrogated survival benefit to PD-1 blockade and leads to development of a Texterm predominant CD8 T-cell population in GL261 that closely mirrors that of CT2A. Finally, we find administration of a CD40 agonist to circumvent CD4 T-cell deficiency is therapeutically synergistic when combined with PD-1 blockade in the CD4 T cell-dysfunctional CT2A model but not in the CD4 T cell-proficient GL261 model.
MethodsAnimals and tumor implantationC57BL/6 female mice (Jackson Laboratory) 8–10 weeks old were acclimated for 2 weeks in specific pathogen-free housing. GL261 was obtained from the National Cancer Institute Tumor Repository, CT2A was obtained from Dr Peter Fecci (Duke University). Tumor cells (5×104) were resuspended in 5 μL phosphate-buffered saline and stereotactically injected into the right striatum of anesthetized mice and randomly distributed into cages representing experimental group, providing age-matched and litter-matched cohorts. Given the nature of subsequent treatments and analyses, the cohorts were not blinded.
Checkpoint inhibitor, CD4 depleting antibody, and CD40 agonist dosingAnti-PDL1 antibody, anti-CD4 depletion antibody, and anti-CD40 agonist were purchased from BioXCell. For scRNA-seq experiments, when indicated, 200 mg CD4 depleting antibody was administered intraperitoneally on day 5 after tumor implantation, and 200 mg of anti-PDL1 was administered intraperitoneally on day 11 after tumor implantation. TIL were harvested on day 14 post implantation. For survival experiments, anti-PDL1 and CD40 agonist were dosed as follows: GL261—CD40 agonist on day 9, anti-PDL1 on days 11, 15, and 19; and CT2A—CD40 agonist on day 7, anti-PDL1 on days 9, 12, 16, and 21 for CT2A. CD4 depletion antibody was administered on day 5 and weekly thereafter. Mortality was recorded or mice were euthanized on reaching predetermined humane endpoints after daily monitoring for neurological deficits, signs of suffering, or impaired function. Kaplan-Meier survival analyses were conducted in GraphPad Prism using Mantel-Cox test. All mice were included for scRNA-seq and survival analyses.
TIL harvesting and T-cell flow cytometric enrichment/analysisFor scRNA-seq experiments, mice were euthanized on postimplantation day 14 (pooled n=3 per model to ensure sufficient cells). Tumors were harvested, dissociated, and filtered through a 70 micron cell strainer. Red blood cells were removed with ACK lysis solution (Thermo Fischer Scientific), and a Percoll density gradient (Sigma-Aldrich) was used for myelin removal. Cell surface staining was performed for live cells (Zombie NIR, BioLegend), CD3 (BioLegend), and CD11b (BioLegend), and cells were sorted on a BD FACSARIAII flow sorter, enriching the live CD3+CD11b− cell population to represent 75% of the total population for further analyses. For intracellular cytokine/transcription factor staining, tumor infiltrating lymphocytes were isolated as previously mentioned then stained using the True-Nuclear Transcription Factor Buffer Kit (BioLegend) with CD45 AlexaFluor700, CD3 PerCP/Cy5.5, CD4 PE/Cy7, CD8 PE Dazzle564, Zombie NIR, PD-1 BV650, Foxp3 BV421 (BioLegend); TCF1 AlexaFluor488 (BD Biosciences); and TOX PE and IL-21 APC (eBiosciences) per manufacturer’s instructions. Quantification of interleukin (IL)-21 was performed by gating on live (Zombie NIR negative) CD3+CD8−CD4+Foxp3− cells. For quantification of TOX and TCF1, gating was on live CD3+CD8+CD4−PD-1+ cells. The absolute number of TIL was determined by performing a cell count on isolated cells using a hemocytometer and then calculating the number of immune subsets based on flow cytometry analysis (ie, for CD8+ TIL=number of cells per sample×%CD45+×%CD3+CD8+). Analysis was performed on FlowJo software V.10.6.0 and statistical analysis was performed on Prism GraphPad.
scRNA-seq library generationA live single-cell suspension of CD3+CD11b- cells were resuspended at 1000 cells/μL in phosphate-buffered saline with 0.04% bovine serum albumin. Single-cell RNA libraries were generated using the Chromium Next GEM Single Cell 5’ library and Gel Bead Kit v1.1; T Cell Receptor (TCR) enrichment was performed with the Chromium Single Cell V(D)J Enrichment Kit for Mouse T cells as per manufacturer’s protocol (10X Genomics). Library quality and concentration were determined using a Bioanalyzer Tapestation (Agilent) for library sequencing on the NovaSeq 6000 instrument at a sequencing depth of ~50K read pairs per cell.
scRNA-seq computational analysesCell Ranger V.3.0.1 (10X Genomics) was used to process, align, and generate feature-barcode matrices against the mouse reference genome. Transcriptomic data were analyzed with the Seurat R package193- V.3.2.3.18 scRNA-seq libraries underwent doublet filtering (cells with greater than 93rd percentile with respect to nCount were removed), and cells with <500 features or >5% mitochondrial content were removed prior to log normalization, cell cycle scoring, and scaling. Dimensionality reduction was performed using PCA and Uniform Manifold Approximation and Projection (UMAP), with principal components selected using the ElbowPlot and JackStraw functions. As previously described, unsupervised cell type inference was performed by training a nearest neighbor algorithm on expression data from mouse Haemopedia.19 20 Cell type predictions were further combined with CD4 and CD8 expression to define whole clusters as either CD4+ and CD8+ T-cell clusters; non-T-cell clusters were not included in subsequent analyses. To address the potential impact of transcript ‘drop-out’ in cell type identification, subsequent CD8+ analyses were repeated with additional rigorous filtering criteria requiring CD8 expression of >0, with no change in the results (data not shown). The CD4+ and CD8+ clusters were individually subsetted and again underwent log normalization and scaling prior to dimensionality reduction for further analyses. Thirty principal components at a cluster resolution of 0.7 were used for CD8 analyses, and 20 principal components at a cluster resolution of 0.9 were used for CD4 analyses. Distributions of UMIs per cell per treatment condition and cell cycle were visualized using the ggplot2 R package. Differentially expressed genes between groups in each cluster were detected using a Wilcoxon rank-sum test from the FindAllMarkers function in the Seurat R package. The expression of the following genes was used to generate a CD8 T-cell exhaustion score using the Seurat package AddModuleScore: LAG3, CTLA4, PDCD1, HAVCR2, TOX, ICOS, TNFRSF4, and TIGIT. Functional enrichment was calculated using the clusterProfiler package.21
V(D)J libraries were processed with CellRanger V(D)J V.3.0.1 (10X Genomics) mapped onto a mouse V(D)J reference genome. Clonotype analysis was performed with the scRepertoire R package V.1.3.1, using the functions quantContig, clonalHomeostasis, clonalProportion, and clonalDiversity.22
ResultsscRNA-seq of TIL from established intracranial murine gliomas reveals a gradient of exhausted CD8 T-cell statesTo study the T-cell populations in murine gliomas with or without PD-1-blockade therapy, mice were implanted intracranially with GL261 or CT2A and allowed to establish over 11 days, at which time half the mice in each group were treated with anti-PDL1 antibody. TILs were harvested 3 days later, and CD3+CD11b− TILs were enriched by flow sorting and submitted for scRNA-seq. The CD8+ population was initially analyzed, and graph-based clustering demonstrated 11 distinct clusters (online supplemental figure S1A). On manual review, clusters 9 and 10 were present exclusively in untreated CT2A and CD4-depleted anti-PDL1 treated GL261, respectively (online supplemental figure S1B). Interestingly, the most highly enriched genes in these clusters were related to the T-cell receptor, suggesting an expanded monoclonal/oligoclonal population may be driving the clustering. Indeed, clonotype analysis demonstrated that these clusters were each composed of monoclonal populations (data not shown). We therefore removed TCR-related genes (online supplemental table 1) and reran scaling, log transformation, and variable gene selection of the CD8 T cells using 31 principal components at a cluster resolution of 0.7. After excluding TCR genes, graph-based clustering of these 5154 cells yielded eight clusters (figure 1A), with redistribution of the expanded oligoclonal cells throughout these clusters (online supplemental figure S1C).
 Figure 1
Figure 1 Single-cell RNA-sequencing characterization of infiltrating CD8+ T cells in six glioma models. (A) UMAP with cells colored according to graph-based cluster on the left (Tex subsets defined in subsequent analyses) and by glioma model on the right. (B) Gene expression dot plot of key genes associated with terminal and progenitor exhausted T-cell (Tex) populations, which defines cluster 1 as Texprog. (C) UMAP and violin plot of left exhaustion module score and right expression of Tcf7. (D) UMAP feature plot of Ki67 expression supports clusters 3 and 7 as a Texprolif subset. Tex, exhausted CD8 T cell; Texprog, progenitor exhausted CD8 T cell; Texprolif, proliferating Tex cell.
We first sought to identify Texprog and Texterm subsets using previously established marker genes, which corresponded to distinct cell clusters, several expressing Texterm markers (clusters 0, 3, 4, and 7) and one expressing Texprog markers (cluster 1) (figure 1B). We generated a composite exhaustion score using Texterm marker genes (see the Methods section) (online supplemental figure S2). This indicated a gradient of exhaustion from cluster 1 (least exhausted) to clusters 0, 3, 4, and 7 (most exhausted) (figure 1C). Consistently, Tcf7, a Texprog marker, was inversely correlated with exhaustion score and predominantly enriched in cluster 1. We then scored each cell for expression of genes associated with the Texterm signature defined in previously published datasets including murine chronic viral and cancer models.7 12 We again noted that clusters 0, 3, 4, and 7 aligned with the Texterm signature (online supplemental figure S3). To identify the cellular processes unique to the four Texterm clusters, we first identified global differentially expressed genes for each cluster, then analyzed their functional enrichment (online supplemental figure S4). Combined with Ki67 expression (figure 1D), this revealed upregulation of cell division and proliferative pathways in clusters 3 and 7, suggesting these clusters represent the previously described proliferating Tex cells (Texprolif), which progressively lose Tcf7 expression as they divide and convert into an irreversible terminally exhausted state.8 Collectively, these data suggest that clusters 0 and 4 represent a Texterm transcriptional state; clusters 3 and 7 align with a Texprolif transcriptional state; and cluster 1 is consistent with a Texprog transcriptional state. Together, these results demonstrate that CD8+ TIL isolated from these two orthotopic glioblastoma mouse models correspond to previously defined CD8 T cell-exhausted states, and the full differentiation gradient of this distinct T-cell lineage is represented.
GL261 and CT2A TILs consist of progenitor and terminally exhausted populations that differentially respond to PD-1 blockade therapy and are mediated by CD4 T cellsWe next assessed the frequency of each Tex subset within CD8+ TIL isolated from GL261 and CT2A tumor-bearing mice. Notably, GL261 was enriched in Texprog, while CT2A was predominantly composed of Texterm (figure 2). In extracranial tumor models, it has been shown that PD-1 blockade therapy reinvigorates Texprog but not Texterm.10 To determine if this holds true in intracranial tumors, we profiled the transcriptional changes induced by PD-1 blockade therapy in GL261 and CT2A. Similar to observations in extracranial tumors,11 12 23 the Texprog enriched GL261 CD8+ TIL demonstrated a shift in transcriptional activity following PD-1 blockade therapy from cluster 1 (Texprog signature) to clusters 2 and 5, while the largely Texterm CD8+ subset of CT2A did not exhibit a substantial transcriptional change (figure 2). To further define the transcriptional changes of CD8+ TIL in GL261 following PD-1 blockade, cluster 5 was analyzed more closely and found to have adopted an early exhausted state with upregulation of several checkpoint molecules (figure 1C). As clusters 2 and 6 demonstrate similar early upregulation of exhaustion markers, these three clusters likely represent an intermediate exhausted CD8 T-cell (Texinter) state. Moreover, the presence of a Texinter is consistent with lineage tracking experiments demonstrating that Texprog CD8 T cells differentiate into Texterm CD8 T cells following checkpoint blockade therapy10 and confirmed in our data by performing a trajectory analysis which placed cluster 5 transitionally between the Texprog and Texterm clusters (online supplemental figure S7). Furthermore, cluster five shows increased expression of genes previously shown to be upregulated in PD-1 blockade-responsive CD8 T cells (online supplemental figure S8).6 9 11
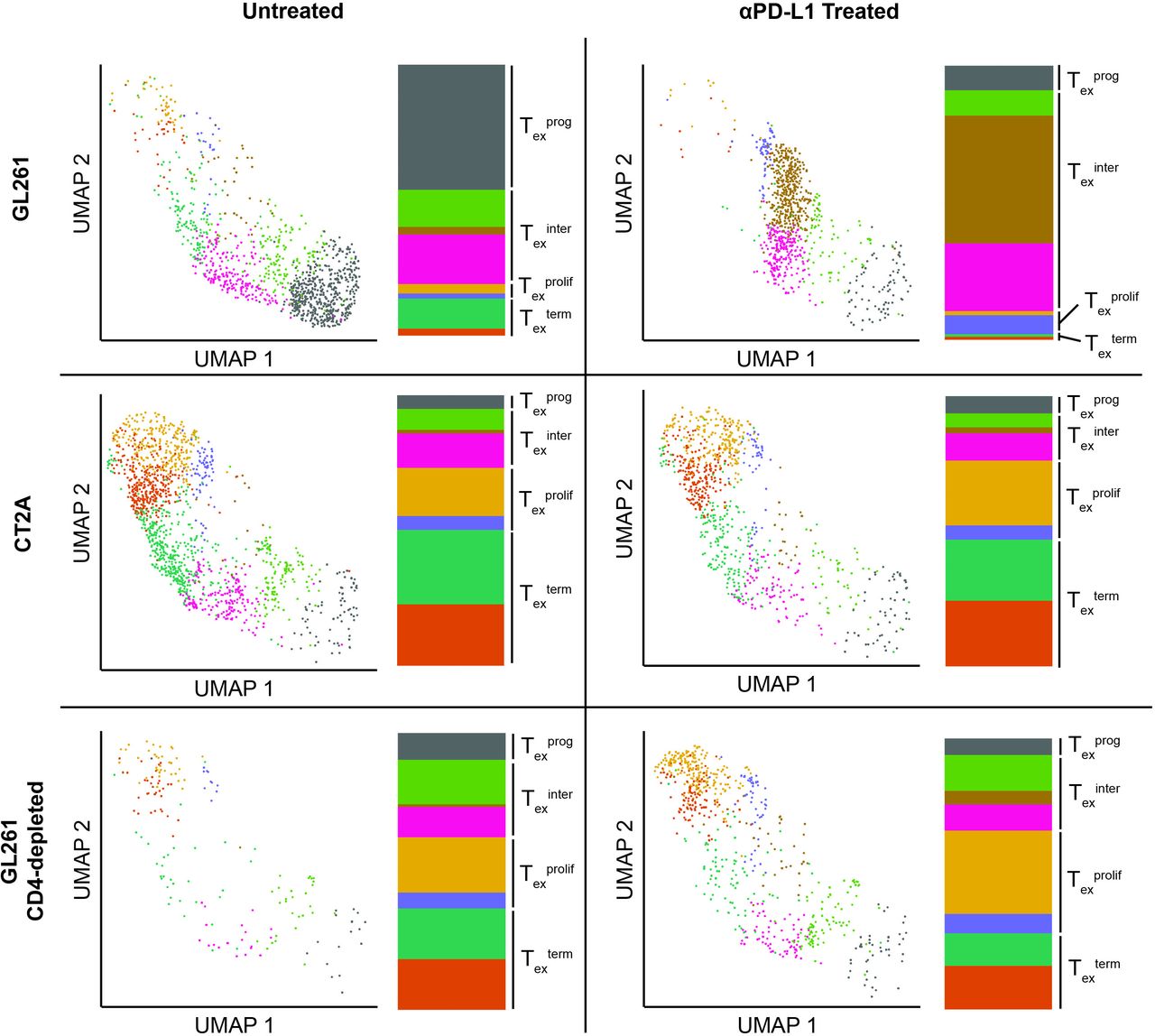 Figure 2
Figure 2 Effect of murine glioma model and PD-1 blockade therapy on CD8+ T-cell compartment. For each model: left UMAP of CD8+ T cells, right histogram of cell composition in each cluster. While GL261 shifts from a Texprog to Texinter predominant model following PD-1 blockade therapy, the CD4-deficient models (CT2A and CD4-depleted GL261) are composed of Texterm cells that are not affected by PD-1-blockade therapy. Tex, exhausted CD8 T cell; Texinter, intermediate exhausted CD8 T cell; Texprog, progenitor exhausted CD8 T cell; Texprolif, proliferating Tex cell; Texterm, terminally exhausted CD8 T cell.
To further validate these transcriptional findings identified between CD8+ TIL in GL261 and CT2A on a protein level, we quantified expression of the Texprog-defining transcription factor, TCF1 (encoded by Tcf7) by flow cytometry. In GL261, approximately 40% of CD8+ TIL are TCF1+, in contrast to only 10% of CD8+ TIL that are TCF1+ in CT2A (figure 3). Conversely, roughly 90% of CD8+ TILs isolated from CT2A express the Texterm-defining transcription factor, TOX, compared with 55% in GL261 (figure 3B). While both GL261 and CT2A contain TOX+CD8+ TIL, the level of TOX expression is significantly lower in GL261 CD8+ TIL than CT2A (figure 3C), though the functional consequence of differences in expression level is unclear. Following PD-1 blockade, there was a significant decrease in TCF1-expressing CD8+ TIL in GL261, similar to what was noted transcriptionally along with a corresponding increase in TOX-expressing CD8+ TIL. In contrast, the percentage of these two populations remained relatively unchanged in CT2A after PD-1 blockade treatment (figure 3A,B), which also paralleled the scRNA-seq data. Thus, the preferential distribution of Texprog in GL261 and Texterm in CT2A is consistent with the observed differential responsiveness of these two models to PD-1 blockade.
 Figure 3
Figure 3 Differences in TCF1 and TOX expression among CD8+ TIL isolated from GL261 and CT2A with and without PD-1 blockade therapy. (A) Bar graph depicting percentage TCF1+PD-1+CD8+ TIL among total PD-1+CD8+ TIL in GL261 and CT2A with and without anti-PDL1 antibody treatment (GL261 TIL vs CT2A TIL, p=0.0025; GL261 TIL vs GL261+PDL1 TIL, p=0.021). (B) Bar graph depicting percentage TOX+PD-1+CD8+ TIL among total PD-1+CD8+ TIL in GL261 and CT2A with and without anti-PDL1 antibody treatment (GL261 TIL vs CT2A TIL, p=0.0003; GL261 TIL vs GL261+PDL1 TIL, p=0.0057). (C) Bar graph representing mean fluorescent intensity of TOX expression in TOX+PD-1+CD8+ TIL (GL261 TIL vs CT2A TIL, p=0.0069). Data pooled from at least two independent experiments with n=3 mice per group per experiment. TIL, tumor-infiltrating lymphocyte.
One potential alternative explanation for differences in responsiveness to PD-1 blockade therapy between these two models is a lack of immune cell infiltration into the CT2A microenvironment compared with GL261. We therefore quantified total numbers of CD8+ and CD4+ TIL among these two models and, in fact, noted an increased number of CD8+ TIL in CT2A compared with GL261 (online supplemental figure S5A), which is supported by the presence of a higher frequency of Texprolif in CT2A (figure 2). Thus, these data suggest that differences in PD-1 blockade responsiveness is not simply due to lack of effector CD8 T-cell infiltration into CT2A.
These data also demonstrate CT2A is not a numerically CD4 lymphopenic tumor microenvironment (online supplemental figure S5A). However, we have previously demonstrated that the CD4+ TILs present in CT2A display a more dysfunctional effector profile than CD4+ TILs from GL261,14 suggesting CT2A may represent a functionally CD4 deplete microenvironment. Given the previously suggested role of CD4 T cells in restraining the development of Texterm CD8 T cells described in chronic Lymphocytic Choriomeningitis virus (LCMV),8 we tested the hypothesis that the response to PD-1 blockade and transcriptional profile of CD8+ TIL in GL261-bearing mice in the setting of CD4 depletion would similarly mirror CT2A. Consistent with this hypothesis, pharmacological depletion of CD4-expressing T cells in GL261 abrogates the survival benefit to PD-1 blockade therapy (online supplemental figure S6) with a corresponding predominance of Texterm CD8+ TIL population unaffected by PD-1 blockade therapy (figure 2). Furthermore, similar to CT2A, CD8+ TIL from CD4-depleted GL261 had an increase in Texprolif (figure 2) and absolute numbers of CD8+ TIL compared with CD4-intact GL261-bearing mice (online supplemental figure S5B). It should also be noted that the relative paucity of Texprog in both CD4-deficient brain tumor models (CT2A-depleted and CD4-depleted GL261) is in slight contrast to the chronic viral infection model, LCMV, where CD4 depletion leads to an increase in Texterm via progressive differentiation of a Texinter population but preservation of the Texprog population.24 Thus, there may a context-dependent role of CD4 T cells on Texprog differentiation influenced by pathology (infection vs tumor) and/or location (intracranial vs extracranial).
In summary, GL261 is composed of Texprog-predominant CD8+ TIL capable of responding to PD-1 blockade therapy, while CT2A-depleted and CD4-depleted GL261 CD8+ TILs represent a Texterm subset insensitive to PD-1 blockade therapy. Together, these findings support the potential role of CD4 T cells to mediate PD-1 blockade therapy efficacy by restricting CD8 T cells from differentiating into an unresponsive Texterm state.
CT2A TIL consists of highly expanded TCR clonotypes, while GL261 has preserved TCR diversityThe emergence of clonally expanded, terminally differentiated CD8 T cells in response to persistent antigen exposure has been described in models of chronic viral infection.7 We therefore analyzed T-cell clonality in these tumor models using the Shannon and Simpson Indices, measures of TCR diversity, which demonstrated GL261 had a more diverse clonotypic repertoire than CT2A (online supplemental figure S9). We ranked clonotypes based on their frequency, assigning a clonal index to each TCR with the dominant clonotype represented by a clonal index of 1 (figure 4). In both tumor models, clonal expansion is higher in the Texterm clusters. This finding is replicated by superimposing clonal groups (defined by degree of expansion) onto the CD8 T-cell UMAP (figure 4B) or the top five clonal indices split by sample (figure 4C). Given the effects of CD4 depletion on the composition of the GL261 CD8+ TIL compartment, we evaluated its effects on TCR heterogeneity and found CD4 depletion in GL261 similarly reduces the clonal diversity near that of CT2A (figure 4A,C).
 Figure 4
Figure 4 Tumor-infiltrating T-cell TCR clonality and diversity (A) TCR heterogeneity of each glioma model, wherein ‘clonal index’ represents each clonotype’s rank-based frequency. (B) Clonal expansion categories superimposed onto UMAP of all conditions concatenated. (C) Visualization of the top five expanded clonotypes of each tumor model superimposed onto individual UMAP.
As proliferation of exhausted Tcf7-expressing progenitor CD8 T cells in response to checkpoint blockade therapy has been described in chronic viral models, we next assessed the influence of PD-1 blockade therapy on the CD8 T-cell compartment in each tumor model.6 As anticipated, given the relative enrichment of Texprog in GL261, we observed clonal expansion in GL261 CD8+ TIL following PD-1 blockade therapy with an accompanying reduction in clonal diversity (figure 4A,C). In contrast, we did not observe these effects in the Texterm predominant CT2A-depleted or CD4-depleted GL261 models (figure 4A,C). Specifically, the untreated GL261 CD8+ TIL compartment was composed predominantly of minimally expanded TCRs, represented primarily by clonal indices of >100 (figure 4B). Conversely, the CD8 T-cell compartments of treated GL261, untreated and treated CD4-depleted GL261, and untreated and treated CT2A all exhibited significant clonal enrichment, with the top 100 clonal indices comprising more than 75% of the TCRs. Notably, each of these hyperexpanded clones appears to be distributed across multiple clusters (figure 4C), suggesting that T cells within a single clonal population exist at various stages of exhaustion. Taken together, while the Texprog-enriched TILs of GL261 undergo a phenotypical and transcriptional transformation with a proliferative burst following PD-1 blockade therapy, no such changes are observed in the dysfunctional CD4 models and may underlie the differential responsiveness of each model to PD-1 blockade therapy.
CT2A is composed of inhibitory regulatory T cells and dysfunctional effector CD4 T cells relative to GL261Given the effect of CD4 T cells on the Tex population in each tumor model, we next analyzed the CD4 T-cell compartment from the untreated GL261 and CT2A TIL, consisting of 1103 cells and 14 013 genes (figure 5). We analyzed the effect of tumor type on the CD4 T-cell compartment; while GL261 (figure 5B) was represented in clusters 0, 1, 3, and 4, the CD4 T-cell compartment of CT2A was primarily enriched in clusters 2 and 3 (figure 5B). We found two clusters (clusters 3 and 4) with high Foxp3 expression indicative of regulatory T cells (Treg) (figure 5C).25 We next compared the expression of genes related to Treg function to delineate resting versus activated states. Tnfrsf9, the costimulatory receptor for tumor necrosis factor family 4-1BB, is a marker of activated Treg, which are capable of increased suppressive capacity of T-cell proliferation and activity.26–28 We found Tnfrsf9 to be highly expressed in the Treg cluster 3, which is enriched in CT2A (figure 5C). Importantly, such activated Treg populations are strongly correlated with poorer prognosis in both systemic and intracranial tumors, along with a lack of response to PD-1 blockade therapy in non-small cell lung cancer.25
 Figure 5
Figure 5 Single-cell RNA sequencing characterization of infiltrating CD4 T cells. (A) UMAP with cells colored according to graph-based cluster. (B) Graph-based clustering and cluster composition of the CD4 T-cell compartment separated by glioma model. (C) Characterization of the regulatory T-cell (Treg) compartment: scaled Foxp3 expression defines clusters 3 and 4 as Treg (left/top); expression dot plot of key Treg genes defines cluster 3 as activated and cluster 4 as resting Treg (right); and histogram of Treg composition shows CT2A is predominantly activated Treg, while GL261 is composed of equivalent proportions of activated and resting Treg (left/bottom). (D) Expression dot plot of effector CD4 T-cell genes in the non-Treg clusters. (E) Expression dot plot of genes comprising the Isc signature, which is more highly expressed in cluster 2.31 (F) Feature plot demonstrating scaled gene expression of IL-21 (left) and bar graph depicting protein expression represented as mean fluorescent intensity of IL-21 within Foxp3− CD4+ TIL (right; data pooled from at least two independent experiments with n=3 mice per group per experiment.; GL261 TIL vs CT2A TIL, p=0.030). IL, interleukin; TIL, tumor-infiltrating lymphocyte; Treg, regulatory T cell.
We next compared the expression of a gene set associated with an exhausted CD4 T-cell state within the Foxp3-negative, effector CD4+ clusters (clusters 0, 1, and 2).29 Cluster 0, enriched in GL261, is marked by elevated expression of key markers of CD4 T-cell exhaustion such as Pdcd1, Ctla4, and Lag3 (figure 5D). This was initially surprising as we anticipated CT2A, enriched in the Texterm subset, would similarly be composed of exhausted CD4 T cells. However, as previously demonstrated in bladder and head and neck cancers, exhausted CD4 T cells do not display the same dysfunction as their exhausted CD8 T-cell counterparts, and in specific contexts, CD4 T cells with an exhausted phenotype represent a checkpoint-responsive subset with the potential to proliferate and increase cytokine production following therapy.30 Gene ontology analysis of the top biological processes demonstrated the CT2A-enriched cluster 2 was associated with response to interferon-responsive genes (online supplemental figure S10), previously correlated with poor response to checkpoint therapy in the murine MC38 adenocarcinoma model.31 Given this association, we quantified expression of a previously described gene signature defining a dysfunctional CD4+ subset characterized by expression of type I interferon (IFN)-responsive genes (Isc) and associated with poor prognosis.29 31 We found the Isc gene signature strongly associated with the CT2A effector CD4 T-cell cluster 2 compared with the GL261 effector CD4 T-cell clusters 0 and 1, further supporting a dysfunctional CT2A effector CD4 T-cell compartment (figure 5E). Finally, given the described role of IL-21 secretion by CD4 T cells in maintaining CD8 T cells in a non-terminal exhaustive state,24 32 we next determined the subpopulations expressing this cytokine, noting restricted expression in the GL261-predominant cluster 0 (figure 5F). This increased expression of IL-21 in CD4 effector TIL from GL261 compared with CT2A was subsequently confirmed on a protein level by flow cytometry (figure 5F). IL-21 was minimally expressed in the other CD4 effector clusters, suggesting a potential mechanism for the disparity of exhausted CD8 T-cell states in GL261 and CT2A.
Thus, scRNA-seq analysis reveals a balanced resting and activated Treg ratio along with activated effector CD4 T cells in the PD-1 responsive GL261 model, which contrasts with the skewing toward an immunosuppressive, activated Treg-enriched and Isc-expressing effector CD4 T-cell subsets in the CT2A model. Importantly, this analysis indicates that a functional CD4 T-cell compartment is key to maintaining PD-1 blockade responsiveness in glioblastoma models.
CD40 agonism synergizes with PD-1 blockade to provide protection in a CD4-dysfunctional but not a CD4-proficient brain tumor microenvironmentOne mechanism by which effector CD4 T cells promote an effective CD8 T-cell antitumor immune response is via CD40–CD40 ligand (CD40L) interactions with dendritic cells to induce upregulation of MHC class I expression and co-stimulatory molecules, and secrete activating cytokines such as IL-12.33 34 Therefore, CD40 agonists are actively being explored as both monotherapy and combinatorial therapies to improve the effector T-cell response in patients with advanced malignancies. Similarly, we hypothesized that a selective deficiency in CD4 T-cell functionality could be bypassed with CD40 agonism. To evaluate this hypothesis, mice with established intracranial CT2A were treated with CD40 agonist with or without PD-1 blockade. No survival benefit was observed with either CD40 or anti-PDL1 monotherapy (figure 6A). However, a significant survival benefit was observed following combinatorial therapy (figure 6), suggesting synergy between these two therapeutic targets. In stark contrast, the addition of CD40 agonist, either as monotherapy or in combination with PD-1 blockade, did not significantly improve survival in GL261-bearing mice (figure 6B). Of note, we delayed the initiation of PD-1 blockade to a later time point resulting in a lower survival rate (figure 6B) similar to what was seen in CT2A. This would allow detection of potential synergy as earlier administration of PD-1 blockade in GL261 has a high cure rate as a monotherapy (online supplemental figure S6). These results support prior findings that the addition of CD40 agonism may augment responsiveness to PD-1 blockade in settings where CD4 T-cell activity is deficient or dysfunctional but not in the context of a proficient effector CD4 T-cell response.
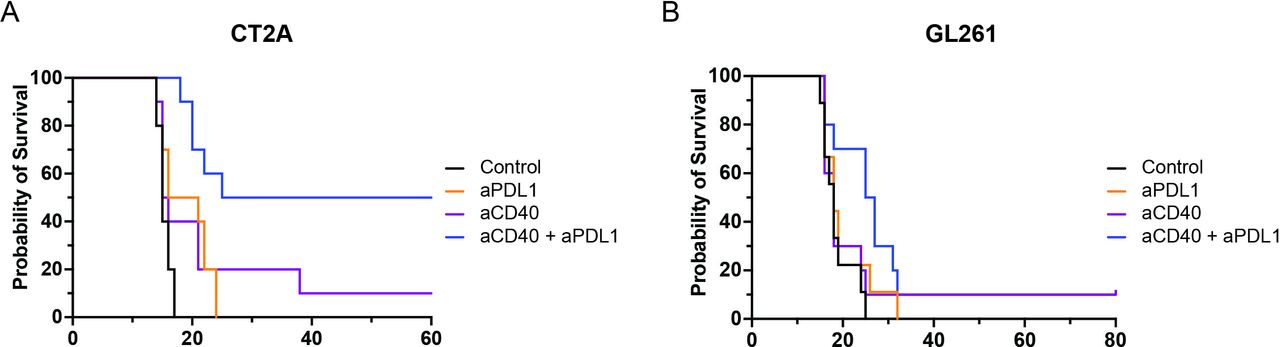 Figure 6
Figure 6 Circumventing a dysfunctional CD4 T-cell response through CD40 agonism. Kaplan-Meier survival curves evaluating CD40 agonist and PD-1 blockade monotherapy and combinatorial therapy in (A) CT2A (p=0.0003) and (B) GL261 (p=0.1129) demonstrate CD40 agonism delivers a synergistic benefit with PD-1-blockade therapy, exclusively in a CD4-deficient environment. For each experiment, n=5 per group and pooled from two independent experiments.
DiscussionStandard of care therapy for primary glioblastoma entails gross total resection followed by concurrent temozolomide and radiation therapy; despite this aggressive regimen, median survival from diagnosis remains less than 2 years.1 Checkpoint blockade therapeutics have achieved durable responses across multiple malignancies. Unfortunately, despite the success of PD-1 blockade therapy to date, this therapeutic modality has yet to demonstrate efficacy in glioblastoma.
A fundamental understanding of treatment failure with checkpoint blockade therapy in patients with glioblastoma could identify strategies to overcome PD-1-blockade resistance and provide additional therapeutic options in an otherwise dismal pathology. One proposed mechanism of glioblastoma resistance to checkpoint blockade is T-cell dysfunction, first described in chronic LCMV infection and more recently demonstrated in murine glioma models.14 For that reason, we leveraged scRNA-seq to explore complementary models of mouse glioblastoma with differing responsiveness to PD-1 blockade therapy, permitting investigation of mechanisms driving therapeutic resistance in brain tumors. We demonstrate presence of a predominant Texterm population associated with CD4-deficient, PD-1 blockade-resistant models (CT2A and CD4-depleted GL261), whereas a predominant Texprog subset is associated with the CD4-proficient, PD-1 blockade-sensitive model (GL261). Importantly, we demonstrate a strategy using CD40 agonism to circumvent a dysfunctional CD4 T-cell response to induce PD-1 blockade responsiveness and provide a survival benefit in an otherwise unresponsive mouse glioblastoma model. Our findings complement prior descriptions of the GL261 and CT2A immunological microenvironment but importantly represent the first application of scRNA-seq to provide a higher-resolution comparative analysis of the CD4 and CD8 T-cell states and clonotypes in these models.14 35
While others have reported a dichotomous split of the CD8 Texprog and Texterm states in models of chronic viral infection, more recently nuanced phenotypical diversity in the CD8 T-cell compartment has been reported by surface markers demonstrating transitional transcriptional states.7 12 23 25 Additionally, shared clonotypes between the CD8 T-cell clusters have previously been demonstrated in systemic tumors and suggest a developmental pathway of exhaustion, further supported by scRNA-seq analyses, suggesting a progressive population between functional and dysfunctional CD8 T cells.25 In fact, this predysfunctional compartment, defined by Tcf7 expression, has been shown to be an integral population for the generation of an antitumor r
留言 (0)